
47 Neurodegenerative Disease: Non-Motor
As we age, the human brain is subject to degradation over our lifetime. This normal aging results in moderate cell loss and a gradual loss of myelination and synapses. These structural changes result in normal age-related functional losses (slower processing time or forgetting where you put your keys).
Alzheimer’s Disease
Alzheimer’s disease was first described In 1907 by Dr. Alois Alzheimer. He had described one of his patients, a 50-year old woman named Auguste Deter, whose symptoms included profound cognitive impairment, memory deficits, and delusions. After Deter’s death, post-mortem analysis of her brain revealed anomalies: degenerating neurons that contained atypical tangles and deposits scattered between cells.
Alzheimer’s disease (AD) is not a natural result of the aging process, but there is an increased risk of AD as we age. It is an irreversible, slowly progressing neurodegenerative condition that leads to deficits in thinking, behavior, and memory loss. AD is a devastating disease that accounts for 60-80% of all cases of dementia and is the sixth leading cause of death in the United States. As for prevalence, approximately 10% of people older than 65 and nearly a third older of people older than 85 has AD.
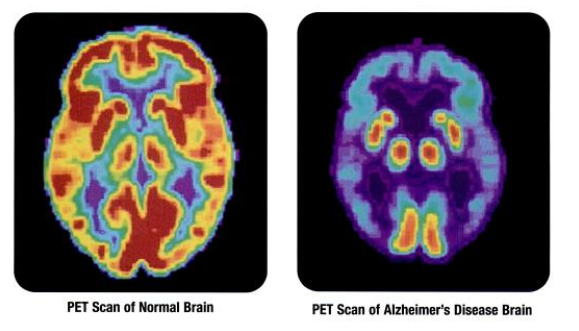
Specifically, declarative memory is the first form of memory loss observed in AD patients. As the disease progresses, procedural memory loss becomes more apparent. In addition to memory loss, AD symptoms include problems with language, disorientation, mood swings, loss of motivation, lack of self care, behavioral problems, loss of bodily functions, and dementia.
AD is divided into two categories, familial and sporadic. Familial AD is diagnosed when a person is in their 50s or 60s, and is strongly influenced by genetic risk factors. This form of AD only makes up about 10% of cases. Sporadic AD is by far more common than familial AD, and is believed to be caused by a combination of old age and environmental factors in addition to genetic risk factors. Several genetic risk factors are linked to AD risk. Apolipoprotein epsilon4 (ApoE4) is the greatest genetic risk factor identified, where individuals who are homozygous for the e4 polymorphism have a 12-times higher risk of developing AD than people with the more common e3 variant. Additionally, mutations in genes like amyloid-precursor protein (APP), presenilin-1 (PSEN1) presenilin-2 (PSEN2), and triggering receptors expressed on myeloid cells 2 (TREM2) are all associated with higher risk of developing AD.
Brain Areas Affected in Alzheimer’s Disease
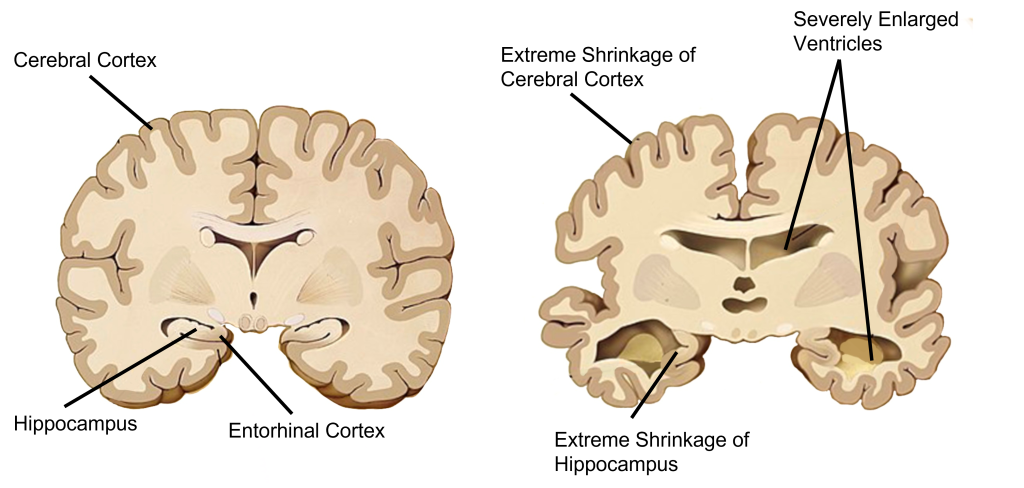
In AD, brain atrophy is observed across the brain, especially in later progression of the disease, however two structures that see the most atrophy are the hippocampus and cortex. One of the main structures affected in Alzheimer’s disease is the hippocampus, which is located curled up inside the temporal lobes. The hippocampus is a critical structure in learning and memory, especially in spatial memory and in consolidation of short-term memory into long-term memory.
The prefrontal cortex is also heavily affected in AD. The prefrontal cortex is the most anterior portion of the frontal lobe and functions in intellect, cognition, recall, problem solving, planning, and personality.
Hypotheses of Alzheimer’s Disease
Pathologically, AD is characterized by the extracellular accumulation of amyloidbeta plaques (Aβ) that build up in the space between neurons and intracellular hyperphosphorylated neurofibrillary tau tangles (NFT).
Beta Amyloid Hypothesis
The amyloid cascade hypothesis, originally proposed in 1992, has become the leading theory in the field describing how AD develops. The hypothesis suggests that the main driving factor of AD is the deposition of Aβ in the brain, and this in turn leads to neurodegeneration via cell death, abnormal protein buildup, and neuroinflammation.
Aβ is produced from the cleavage of amyloid-precursor protein (APP), an integral membrane protein normally expressed by neurons that typically functions as a cell surface receptor and is important in neuronal adhesion, neurite growth, and axogenesis. In AD, the extracellular soluble portion of APP is cleaved by the enzyme beta-secretase and then the enzyme gamma secretase cleaves the APP anchor, leaving Aβ protein that is no longer held within the membrane. Aβ is likely to clump together in unpredictable ways within the extracellular space, leading to the formation of Aβ plaques (pronounced “A beta”). These plaques can cause neuronal death and lead to the cognitive deficits observed in AD patients.
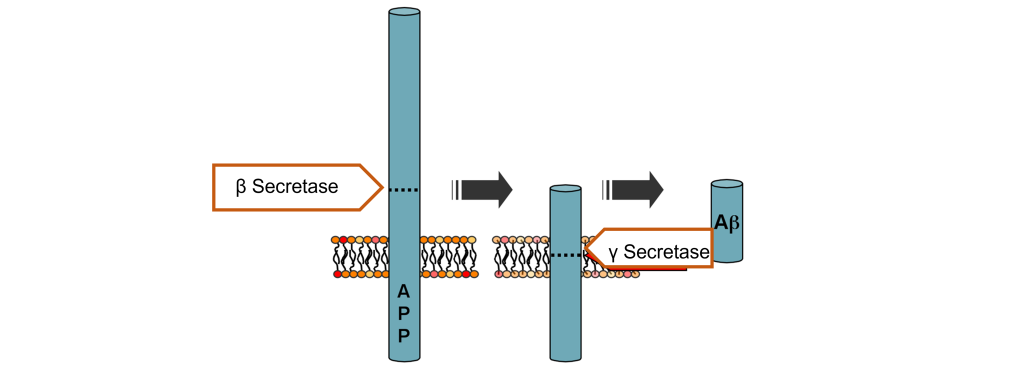
Two major genetic risk factors for AD, presenilin 1 and 2, are mutations of the gamma secretase that leads to increased APP cleavage. Therapies are being developed to target and control the Aβ protein to mitigate AD symptoms. Recently an antibody-based drug has been approved by the FDA. This groundbreaking drug is unique from those traditionally prescribed because it directly targets Aβ. However, the amyloid-cascade hypothesis does not tell the whole story. For one, there are patients who carry a heavy Aβ load but present no clinical symptoms of AD. More so, in mouse models with APP mutations, they develop significant Aβ plaques, but have no accumulation of tau (see below) and no significant neurodegeneration. Additionally, there have been several drugs targeting Aβ that have failed in human trials.
Tau Tangle Hypothesis
While these amyloid plaques seem to be the primary driving factor of AD, hyperphosphorylated tau and neurofibrillary tangles (NFTs) are also potential culprits in disease progression. NFTs are an intracellular pathological marker of AD and correlate strongly with cognitive deficits in AD. Tau protein is the major microtubule-associate protein (MAP) of mature neurons and helps to function to maintain cellular morphology. Excess phosphorylation of this protein causes tau to accumulate inside the cell, leading to neuronal dysfunction and cell death. The presence of Aβ increases the levels of the tau, and vice versa, adding to the complexity and difficulty of treating AD. Tau pathology is also observed in other neurodegenerative diseases such as frontotemporal lobe dementia and Parkinson’s disease.
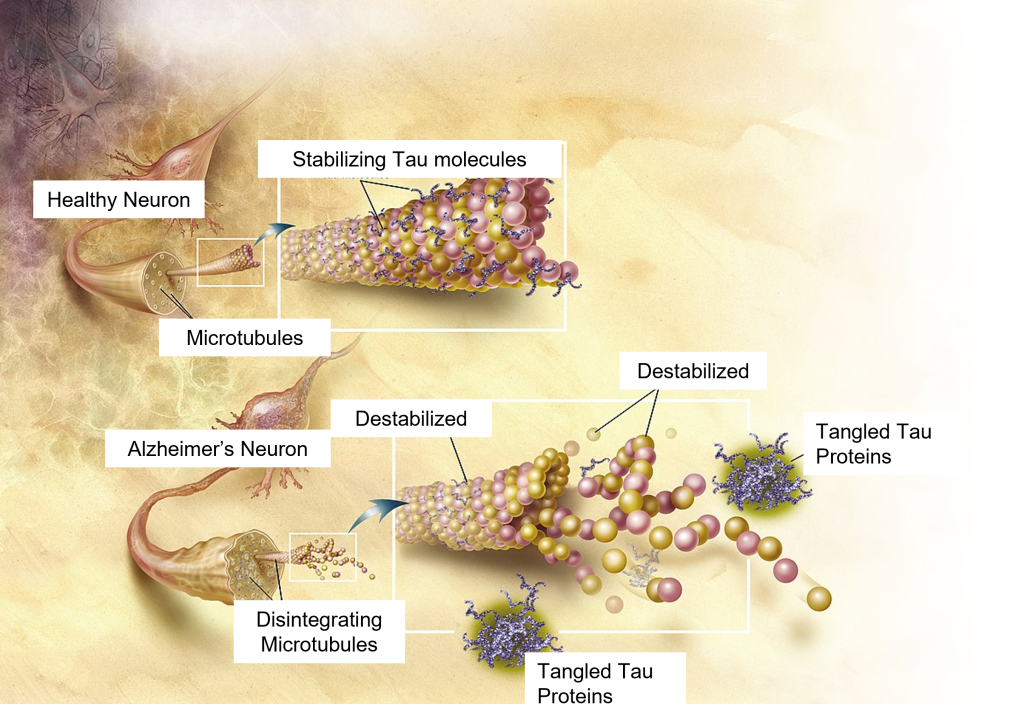
Although most discussion of AD revolves around plaques and tangles, it is well known that a cacophony of pathological markers are also seen in AD, including neuroinflammation, oxidative stress, blood-brain barrier dysfunction, heavy metal dysregulation, mitochondrial impairment, and many more.
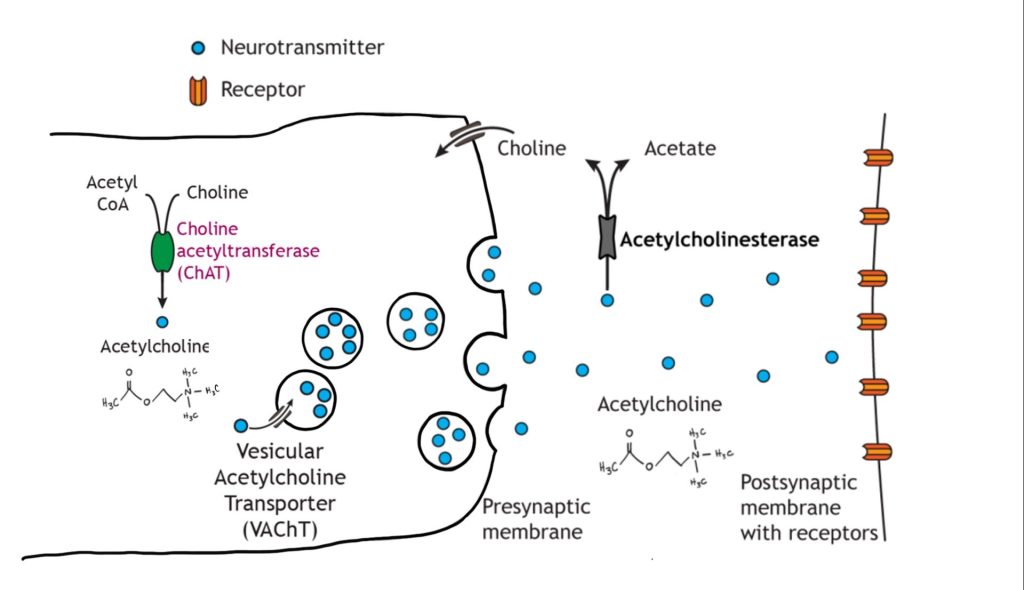
Acetylcholine Hypothesis
An early hypothesis proposed to explain the neuronal loss and memory deficits observed in AD is centered around changes in the signaling of acetylcholine. Early post-mortem analyses discovered that people with AD have profound atrophy of the basal forebrain, an area which contains a large density of cholinergic neurons. Further, patients with Alzheimer’s disease also show decreased levels of choline acetyltransferase, reduced choline uptake in cells, and reduced release of acetylcholine. The cholinergic hypothesis suggests that it is this loss of cholinergic neurons and the loss of acetylcholine signaling that is the main pathological driver of AD. The theory is supported by the idea that acetylcholine plays an important role in learning and memory.
Diagnosis of Alzheimer’s Disease
To this day, only post-mortem analysis can conclusively diagnose someone with AD. As expected, this raises many issues in terms of treating these patients. Currently, physicians diagnose a patient with AD based on the symptoms they presented with. However, new techniques to make more accurate diagnoses are emerging, including identifying the presence of biomarkers from blood samples, certain functional characteristics as observed in PET or fMRI imaging, and possibly even through eye exams.
Mild cognitive disorder/impairment represents a modest level of cognitive decline that interferes with the ability to complete everyday activities. Whereas major cognitive disorder/impairment is considered a major impairment and includes different dementias and Alzheimer’s disease.
Treatments
Most of the FDA approved drugs for AD are acetylcholinesterase (AChE) inhibitors, drugs that act to increase levels of acetylcholine. The other approved FDA drug is an NMDA receptor antagonist. This drug is prescribed because it is hypothesized that AD may involve glutamate excitotoxicity, where excess binding of glutamate can increase intracellular Ca2+ levels leading to Ca2+ poisoning and neuron death.
Unfortunately, these compounds show only short-term benefits for cognitive function, and these therapeutic effects subside over time. The different drugs are prescribed during different stages of AD, have modest effects and have unwanted side effects.
Key Takeaways
- Alzheimer’s disease is not a normal result of again, but is a neurodegenerative disease that is more likely to affect older adults.
- The cortex and hippocampus are both heavily affected in Alzheimer’s disease, resulting in brain atrophy.
- There are three hypotheses for the cause of Alzheimer’s disease:
- The beta amyloid hypothesis involves beta amyloid plaques that form in the extracellular spaces around neurons, ultimately causing cell death.
- The neurofibrillary tau tangle hypothesis involves destabilization of the tau protein inside of neurons, resulting in the collapse of cellular structure and aggregation of tau proteins into tangles within the cell that cause neuron death.
- The acetylcholine hypothesis stems from observations that acetylcholine signaling is decreased in Alzheimer’s disease.
- Absolute diagnosis of Alzheimer’s disease occurs post-mortem. Physician examinations can make a probable diagnosis prior to death.
- Treatments for Alzheimer’s disease include acetylcholinesterase inhibitors and NMDA receptors antagonists.
Attributions
Portions of this chapter were remixed and revised from the following sources:
- Open Neuroscience Initiative by Austin Lim. The original work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
Media Attributions
- PET_scan-normal_brain-alzheimers_disease_brain © Health and Human Services Department, National Institutes of Health, National Institute on Aging adapted by Valerie Hedges is licensed under a Public Domain license
- Structural differences © Garrondo adapted by Valerie Hedges is licensed under a Public Domain license
- APP Processing © Ipeltan adapted by Valerie Hedges is licensed under a Public Domain license
- Tau Tangles © ADEAR: "Alzheimer's Disease Education and Referral Center, a service of the National Institute on Aging." adapted by Valerie Hedges is licensed under a Public Domain license
- Acetylcholine signaling © Casey Henley adapted by Valerie Hedges is licensed under a CC BY-NC-SA (Attribution NonCommercial ShareAlike) license
- Superior-pattern-processing-is-the-essence-of-the-evolved-human-brain-fnins-08-00265-g0004 © Mattson M adapted by Valerie Hedges is licensed under a CC BY (Attribution) license
irreversible, slowly progressing neurodegenerative condition that leads to deficits in thinking, behavior, and memory loss
memory that involves remembering events and facts. Also called explicit memory.
Type of unconscious memory that allows for remembering procedures and performance of specific tasks
memory that allows for remembering different locations and spatial relationships between things
in front of; toward the face
intracellular pathological marker of Alzheimer's disease made up of protein Tau

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}