
Chapter 7: Nucleophilic attack at the carbonyl carbon:
There is a set of organic compounds that incorporates the carbonyl group (C=O) which includes aldehyde ketones, carboxylic acids, and carboxylic acid derivatives such as: esters, amides, acid anhydrides, and acid chlorides (as shown in Table 7.1).
Table 7.1: Functional groups that contain a carbonyl group
| Functional Group | Example | Name |
| Aldehyde | 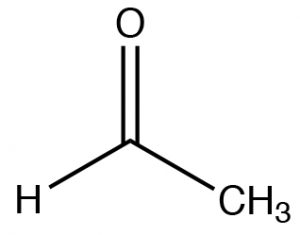 |
Longest chain with aldehyde:
remove -ane, add -al Ethanal (IUPAC)[1] Acetaldehyde |
| Ketone | 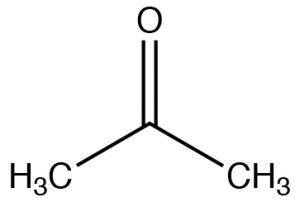 |
Longest chain: remove -e, add -one
2-propanone (IUPAC) Acetone |
| Carboxylic Acid | 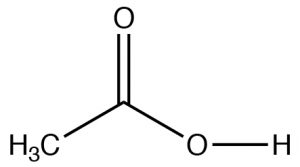 |
Longest chain with CO2H: remove -e, add -oic acid
Ethanoic acid (IUPAC) Acetic acid |
| Ester | 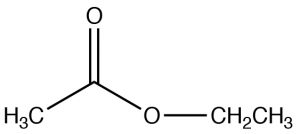 |
Named as a derivative of the CO2H, remove -icacid and -ateEthyl ethanateEthyl acetate |
| Amide | 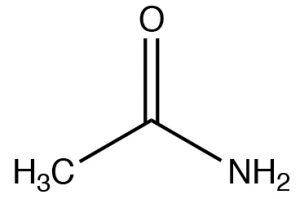 |
Named as a derivate of the CO2H, remove -icacid, add -amideEthanamideAcetamide |
| Acid chloride | 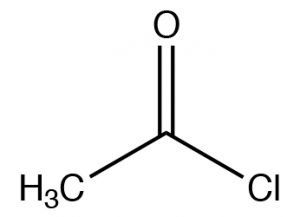 |
Named as a derivate of the CO2H, remove -icacid, add -yl chloride (or other halide)Ethanoyl chlorideAcetyl chloride |
| Acid anhydride | 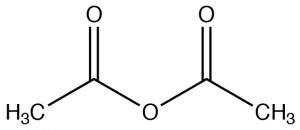 |
Named as a derivative of the CO2H, remove “acid” and add “anhydride”
Ethanoic anhydride Acetic anhydride |
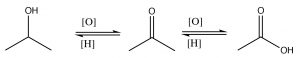 These functional groups have a number of similarities and some notable differences in their properties, which can be predicted on the basis of our understanding of structure and function relationships. These carbonyl compounds can be classified into two broad groups: 1) ketones and aldehydes and 2) carboxylic acids and derivatives (amide, chloride, ester, and anhydride). These two, broad groups differ in the oxidation level of the carbonyl carbon: aldehydes and ketones have two bonds to the electronegative oxygen; acids and derivatives have three bonds to electronegative atoms (O, N or Cl); and, of course, alcohols only have one bond. Interconverting between alcohols, ketones, and carboxylic acids involves some kind of redox reaction. We have already discussed the alcohol to ketone (or aldehyde) transformation and, later, we will discuss further oxidation to the acid level.
These functional groups have a number of similarities and some notable differences in their properties, which can be predicted on the basis of our understanding of structure and function relationships. These carbonyl compounds can be classified into two broad groups: 1) ketones and aldehydes and 2) carboxylic acids and derivatives (amide, chloride, ester, and anhydride). These two, broad groups differ in the oxidation level of the carbonyl carbon: aldehydes and ketones have two bonds to the electronegative oxygen; acids and derivatives have three bonds to electronegative atoms (O, N or Cl); and, of course, alcohols only have one bond. Interconverting between alcohols, ketones, and carboxylic acids involves some kind of redox reaction. We have already discussed the alcohol to ketone (or aldehyde) transformation and, later, we will discuss further oxidation to the acid level.
As we discussed in Chapter 6, the carbonyl carbon is highly polarized; the large [latex]\sigma[/latex]+ on the carbon makes it susceptible to nucleophilic attack. There are a large number of reactions that begin by the attack of a nucleophile on a carbonyl group. To make understanding these reactions more manageable (intelligible), we will consider these reactions in a sequence of increasing complexity, beginning with reactions of aldehydes and ketones.[2] We will then cycle back around and visit similar reactions involving acids and their derivatives.
Aldehydes and Ketones
Is there a difference in reactivity between aldehydes and ketones? Not really; both types of compounds undergo nucleophilic attack, although, in general, aldehydes react faster than ketones for two reasons:
- Aldehydes are less hindered at the carbonyl carbon than ketones (since there is always at least one H attached whereas ketones always have two bulkier alkyl groups).
- Alkyl groups are electron-donating so the partial positive charge on the carbon is partially offset by induction from the alkyl groups.
In practice, however, there is generally little difference between the two and so unless we point out a difference you can assume that they undergo similar reactions.
The similarities between aldehydes and ketones are supported by an examination of their spectra. For example, both aldehydes and ketones display a strong carbonyl absorption around 1700 cm–1. As we will see shortly, not all carbonyls absorb in this area—their C=O stretching frequencies are dependent on their electronic environment. In C-13 NMR, both aldehydes and ketones show a low field peak for the C=O around 200 ppm indicating that the electronic environment of the carbonyls are similar. The major difference in the spectra is the aldehyde proton resonance in the HNMR that appears downfield. This peak often appears as a singlet, even though it may be adjacent to a C-H group, because the coupling constant is often small and, depending on the sensitivity of the instrument used, splitting may not be detectable.
Nucleophilic attack by hydride or carbanions
As we discussed in Chapter 6, aldehydes and ketones react with reagents that are able to deliver hydride (for example from sodium borohydride) or a carbanion (in the form of a Grignard reagent) to the carbonyl group.
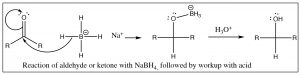
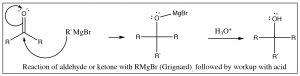
In addition to Grignard reagents, carbanions can also be generated by treating alkyl halides with lithium under dry conditions. These alkyl lithium reagents (RLi) behave in a very similar way to Grignard reagents, although they are somewhat more reactive.
These two reactions: addition of hydride (reduction) or a carbanion (resulting in carbon-carbon bond formation) to a carbonyl are analogous. They also have something else in common in that, under normal laboratory conditions, they are not reversible because reversing the reaction would require that the hydride ion or carbanion be expelled from the central carbon. These species are very unstable, very strong bases. The reagents that produced them (NaBH4, RMgX, RLi) are specialized reagents that do not contain “naked” hydride ions or carbanions. If we wanted to reverse them, we would have to use completely different reaction conditions that avoided the expulsion of the high-energy hydride or carbanion. For example, to accomplish the reverse of the reduction reaction, we would have to use an oxidizing agent (such as Cr(VI)) under completely different conditions. 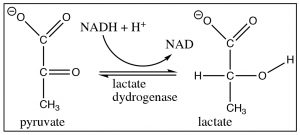 The reduction of a ketone group is central to the reactions of glycolysis, in which pyruvate (the conjugate base of pyruvic acid[3]) is reduced to lactate (the conjugate base of lactic acid) by NADH (see Chapter 6). This reaction can be also reversed under different conditions (in this case, the presence of the enzyme lactate dehydrogenase). Glycolysis will be discussed in more detail in Chapter 9.
The reduction of a ketone group is central to the reactions of glycolysis, in which pyruvate (the conjugate base of pyruvic acid[3]) is reduced to lactate (the conjugate base of lactic acid) by NADH (see Chapter 6). This reaction can be also reversed under different conditions (in this case, the presence of the enzyme lactate dehydrogenase). Glycolysis will be discussed in more detail in Chapter 9.
Reaction with other carbanions: There are a number of other ways to generate carbanions: for example, terminal alkynes are quite acidic (pKa 22) and can be deprotonated by sodium amide (Chapter 5). 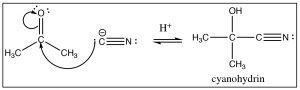 The resulting carbanion adds to the carbonyl just as we might expect. Similarly, cyanide ion (CN–) is another source of a negatively charged carbon. It is a good nucleophile, and just as one might expect, it adds to carbonyl groups, and after reaction with a dilute acid, the resulting cyanohydrin is formed. There are two items to note here:
The resulting carbanion adds to the carbonyl just as we might expect. Similarly, cyanide ion (CN–) is another source of a negatively charged carbon. It is a good nucleophile, and just as one might expect, it adds to carbonyl groups, and after reaction with a dilute acid, the resulting cyanohydrin is formed. There are two items to note here:
- Sodium cyanide NaCN (the usual form of cyanide ion) is highly toxic, so don’t try this at home.
- The oxidation state of the carbon in the cyano group is the same as a carboxylic acid. As we will see later, this reaction will come in very useful. All of these reactions are the result of a nucleophilic addition to the carbonyl group, during the course of which the carbonyl carbon rehybridizes from sp2 to sp3.
Reactions of Aldehydes and Ketones with Oxygen Nucleophiles
In contrast to the addition of hydrogen or carbon nucleophiles, the addition of oxygen and nitrogen nucleophiles is reversible under the conditions in which the reaction occurs. This is because (as we will see) the addition of an oxygen or nitrogen nucleophile results in a tetrahedral intermediate that can regenerate the carbonyl by expelling a leaving group. In contrast to the cases with carbon or hydrogen nucleophiles, oxygen and nitrogen nucleophiles can be good leaving groups. Typically, the reaction is catalyzed either by acid or base as discussed below. For example: in aqueous solution, most aldehydes and ketones will react with water to produce a hydrate.
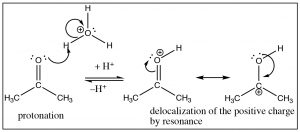 In acid, the first step is protonation of the carbonyl oxygen, the resultant positive charge is partially delocalized onto the carbon [latex]\rightarrow[/latex], which makes the carbonyl more susceptible to nucleophilic attack even by a relatively poor nucleophile such as water.
In acid, the first step is protonation of the carbonyl oxygen, the resultant positive charge is partially delocalized onto the carbon [latex]\rightarrow[/latex], which makes the carbonyl more susceptible to nucleophilic attack even by a relatively poor nucleophile such as water.
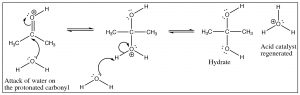
This reaction is completely reversible in aqueous solution, and if any aldehyde or ketone is dissolved in water there is always some equilibrium concentration of the hydrate. In fact, formaldehyde (H2C=O) exists almost exclusively as the hydrate in aqueous solution, whereas most other aldehydes and ketones exist mainly in the carbonyl form. However, it is important to keep in mind that both forms are typically present, and therefore further reactions can proceed from either the hydrate or the carbonyl form.
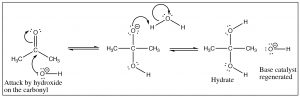
Base catalysis differs in that the first step is attack by the hydroxide (rather than water) on the carbonyl. Since hydroxide is more reactive than water, the carbonyl does not need to be activated by protonation. What the two mechanisms have in common is the rapid protonation/deprotonation reactions that take place in the intermediate steps. We have already seen this many times and with the reactions of aldehydes and ketones, it becomes more important to appreciate just how ubiquitous protonation and deprotonation are. By controlling the pH or the amounts of reactants or products we will see that it is possible to direct such reactions so that the product we desire is produced.
The hydrate is an example of a structure that will play a major part in our discussion of all carbonyl compounds—which we will refer to as the “tetrahedral intermediate.” In this tetrahedral form, the carbon is in the same oxidation state as the ketone (two bonds to oxygen) but in a different hybridization state, sp3 for the hydrate and sp2 for the carbonyl. In most cases, the C=O bond (in the sp2 hybridized form) is stronger (745 kJ/mol in a typical ketone) than two single C–O bonds (2 x 358 kJ/mol) (in the sp3 hybridized form), which explains why (when there is a low-energy stable leaving group) the tetrahedral intermediate that is formed by attack on the sp2 carbon usually collapses back down to a C=O, expelling a leaving group at the same time. As we move forward, we will see the move from tetrahedral to C=O many times, the difference in many of the reactions is which group will leave during this process.
If we change the solvent to an alcohol, we see that the same type of reaction occurs. The alcohol oxygen attacks the carbonyl carbon, but we find that the reaction proceeds further. The first product, formed by addition of one alcohol to the carbonyl is called a hemiacetal but then the reaction continues. Each step is reversible (with low activation energy), each protonation and deprotonation is reversible. All of the oxygens in the molecule can be protonated and deprotonated. When the OH group of the hemiacetal is protonated, it is turned into a good leaving group (H2O) and the carbon undergoes another attack by an alcohol molecule. The end result is an acetal[4] and water.
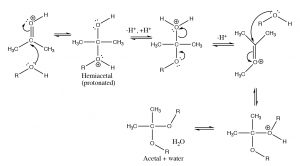
Since these reactions are reversible, you may be asking how we can control the reaction. In this case we can use Le Châtelier’s principle: we can either add a lot of starting material or we can remove one of the products as it is formed. In this case, acetal formation is usually done using the alcohol as solvent, and the water that is formed is removed, so that the position of equilibrium is shifted over to produce the acetal. However, as we will see shortly, sometimes we want to regenerate the carbonyl compound, and this can be done by adding water (and acid catalyst) so that the equilibrium shifts back.
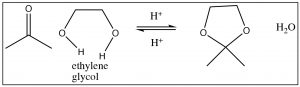 If we use a diol such as ethylene glycol (OHCH2CH2OH), the resulting cyclic acetal is formed. This is frequently used to protect carbonyl groups in more complex molecules, for example, if we wanted to do a reaction in another part of the molecule. Again, the carbonyl group is easily regenerated. The mechanism of hydrolysis is simply the reverse of the acetal formation, beginning with protonation, attack by water, and so on as shown below.
If we use a diol such as ethylene glycol (OHCH2CH2OH), the resulting cyclic acetal is formed. This is frequently used to protect carbonyl groups in more complex molecules, for example, if we wanted to do a reaction in another part of the molecule. Again, the carbonyl group is easily regenerated. The mechanism of hydrolysis is simply the reverse of the acetal formation, beginning with protonation, attack by water, and so on as shown below.
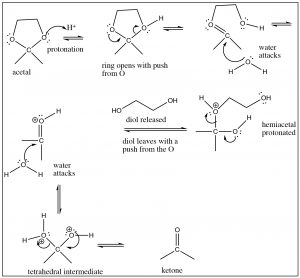
While this mechanism may look (a bit) complicated, in fact, each step is simple and we have seen similar things many times. The issue with these kinds of reactions is that all the oxygens are being protonated and deprotonated all the time. 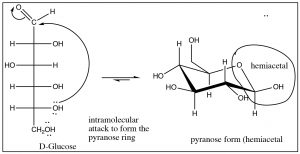 Note that the ways that tetrahedral intermediates behave depends on which oxygen is protonated, and which nucleophile (water or alcohol) attacks the protonated acetal or ketone.
Note that the ways that tetrahedral intermediates behave depends on which oxygen is protonated, and which nucleophile (water or alcohol) attacks the protonated acetal or ketone.
One common example of hemiacetal formation is the intramolecular cyclization of D-glucose to form a six-membered ring which contains a hemiacetal group (among many others). This case is actually a rare example of the tetrahedral form (hemiacetal) being more stable than the carbonyl form.
Reactions with Nitrogen Nucleophiles
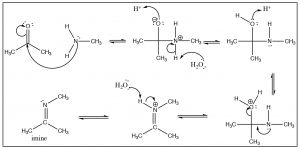 There is a similarity between the reactions of oxygen nucleophiles and nitrogen nucleophiles. For example, let us look at the reaction of a primary amine with a ketone. The mechanism begins in the same way with the nucleophile(N) attacking the carbonyl to form a tetrahedral intermediate, which can undergo various reversible protonation/deprotonation reactions until an intermediate is formed that can collapse down to a new product with a C=N function—which is called an imine. It is the nitrogen analog of the ketone and behaves in much the same way.
There is a similarity between the reactions of oxygen nucleophiles and nitrogen nucleophiles. For example, let us look at the reaction of a primary amine with a ketone. The mechanism begins in the same way with the nucleophile(N) attacking the carbonyl to form a tetrahedral intermediate, which can undergo various reversible protonation/deprotonation reactions until an intermediate is formed that can collapse down to a new product with a C=N function—which is called an imine. It is the nitrogen analog of the ketone and behaves in much the same way.
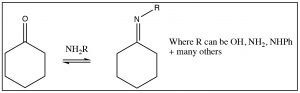 There are many other nitrogenous nucleophiles that can react with aldehydes and ketones, for example hydroxylamine (NH2OH), or hydrazine (NH2NH2) or a whole range of substituted hydrazines, all react with aldehydes and ketones to produce the corresponding imine.
There are many other nitrogenous nucleophiles that can react with aldehydes and ketones, for example hydroxylamine (NH2OH), or hydrazine (NH2NH2) or a whole range of substituted hydrazines, all react with aldehydes and ketones to produce the corresponding imine.
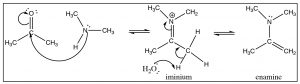 Generally, we do not see the nitrogen analog of an acetal, the intermediate is unstable and reacts to form the imine. However, in the case of a secondary amine, the reaction proceeds in exactly the same way until the imine stage—with the one difference that the nitrogen is now in the quaternary state (an iminium ion). Instead of addition of another amine, a proton is removed to produce a new functionality, the enamine.
Generally, we do not see the nitrogen analog of an acetal, the intermediate is unstable and reacts to form the imine. However, in the case of a secondary amine, the reaction proceeds in exactly the same way until the imine stage—with the one difference that the nitrogen is now in the quaternary state (an iminium ion). Instead of addition of another amine, a proton is removed to produce a new functionality, the enamine.
A note on how these reactions proceed: While these mechanisms may seem complex with many steps, those individual steps are very similar. Students often ask how they can know which way the reaction goes, and the way we write out mechanisms does tend to give the idea that each step is marching purposefully along like clockwork—from one intermediate to another as if the molecules had an “end goal” in sight. Nothing could be further from the truth: each step in the reaction, each protonation and deprotonation, and each nucleophilic attack and loss of a leaving group is occurring all at the same time in a stochastic and chaotic fashion. However, we can control the reaction as discussed earlier by using Le Chatelier’s principle[5]: adding reactants or removing products can shift the position of equilibrium to produce the product that we are after.
Carboxylic Acids and Derivatives
Now that we have a fairly solid understanding of the reactions of aldehydes and ketones, we are going to move up one oxidation state to look at the behavior of carboxylic acids and their derivatives (Table 7.1), a group of compounds that includes the acids, esters, amides, acid chlorides, and acid anhydrides. Just as we did with aldehydes and ketones, we will highlight and discuss the reasons for both the similarities and differences observed. The most obvious difference between this group of compounds is that the carboxylic acids are acidic; the other derivatives lack an acidic hydrogen bonded to an O and, therefore, do not participate in simple acid-base reactions[6]. Since we have discussed the reasons for the acidity of carboxylic acids earlier, we will not go over that here at great length, but be sure to check Chapter 1 if you need a refresher. However, we do want to remind you that many organic compounds are acidic (or basic) and can exist as their conjugate base (or acid) in aqueous solutions, and that the relative amounts of conjugate acid or bases change as pH changes. The degree to which a molecule exists in an acidic or basic form (in water) is particularly important for biological systems that (in humans) are buffered at around 7.3–7.4. Recall that we can relate the pH of a buffered solution to the pKa of any acid that is participating in the solution using the Henderson-Hasselbalch equation[7]:
pH = pKa + log [latex]\frac{[A–]} {[HA]}[/latex],
where HA and A– are the concentrations of the acid and its conjugate base, respectively. We can rewrite this equation (by taking the anti-log of the terms) so that:
[latex]\frac{[A–]} {[HA]}[/latex]= 10(pH-pKa)
which allows us to estimate the relative amounts of acid and base.
For example, a typical carboxylic acid has a pKa of around 4. At physiological pH (~7), the ratio of the conjugate base to conjugate acid is ~10(7-4) = 103, that is, there is about 1000 times more of the conjugate base than the conjugate acid for most common carboxylic acids in biological systems.
There are many naturally occurring (that is, biologically relevant) carboxylic acids and most of them exist as the conjugate base at physiological pH. One consequence is that these species are soluble in water because of the favorable ion-dipole interactions that can be formed. Biological molecules that do not contain such polar (ionized) groups (-COO– or –NH3+) are typically insoluble in water: indeed, the presence of this type of polar (ionic) side chain explains the water solubility of many large biological molecules.
Infra-red spectra as evidence of carboxylic acid derivative structure
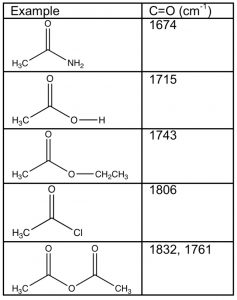 Carboxylic acids and their derivatives have a number of features in common, the most obvious being that the carbonyl carbon has three bonds to electronegative elements—the carbonyl carbon is, therefore, in a higher oxidation state than aldehydes and ketones. However, the nature of the electronegative atom bonded to the carbonyl does impact the properties of the molecule as a whole. For example, if we examine the carbonyl absorptions of these derivatives, we find quite a wide range of frequencies. Recall that most aldehydes and ketones absorb around 1710-30 cm–1, similar to the value displayed by carboxylic acids. Derivatives of carboxylic acids, however, range from around 1670 cm–1 for amides to 1810-30 cm–1 for acid chlorides and anhydrides. How can we explain the difference and use it to predict (and explain) differences in the properties of these derivatives?
Carboxylic acids and their derivatives have a number of features in common, the most obvious being that the carbonyl carbon has three bonds to electronegative elements—the carbonyl carbon is, therefore, in a higher oxidation state than aldehydes and ketones. However, the nature of the electronegative atom bonded to the carbonyl does impact the properties of the molecule as a whole. For example, if we examine the carbonyl absorptions of these derivatives, we find quite a wide range of frequencies. Recall that most aldehydes and ketones absorb around 1710-30 cm–1, similar to the value displayed by carboxylic acids. Derivatives of carboxylic acids, however, range from around 1670 cm–1 for amides to 1810-30 cm–1 for acid chlorides and anhydrides. How can we explain the difference and use it to predict (and explain) differences in the properties of these derivatives?
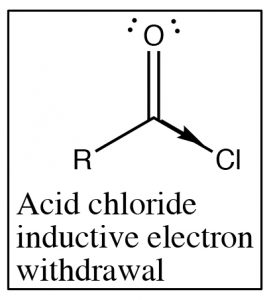 Recall that the IR absorption frequency of carbonyls depends on the energy required to stretch the bond (which is determined by the bond energy). Since the amide absorption frequency is much lower than the absorption frequency of the acid chloride we can conclude that the carbonyl group in the amide requires less energy to stretch than the acid chloride (the bond is weaker). If we look at the structures of these two functional groups we see that both involve an electronegative element bonded to the carbonyl carbon, leading to electron withdrawal by induction through the sigma bond to the carbon.
Recall that the IR absorption frequency of carbonyls depends on the energy required to stretch the bond (which is determined by the bond energy). Since the amide absorption frequency is much lower than the absorption frequency of the acid chloride we can conclude that the carbonyl group in the amide requires less energy to stretch than the acid chloride (the bond is weaker). If we look at the structures of these two functional groups we see that both involve an electronegative element bonded to the carbonyl carbon, leading to electron withdrawal by induction through the sigma bond to the carbon. 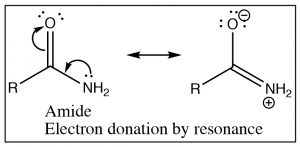 The difference between the two (amide and acid chloride) arises because the amide nitrogen can also donate its electron pair to the carbonyl oxygen. The result is that in amides there is significant overlap between the lone pair of the amide nitrogen and the carbonyl pi bond system. The C-O bond now has less double bond character and therefore it takes less energy to stretch. In contrast chlorine (or any halogen) is not basic and so does not participate in this kind of resonance through the pi system. In acid chlorides, the chlorine is removing electron density from the carbon by induction through the pi system; there is more C=O double bond character than in an amide and it takes more energy to stretch the carbonyl bond (leading to a higher IR absorption).
The difference between the two (amide and acid chloride) arises because the amide nitrogen can also donate its electron pair to the carbonyl oxygen. The result is that in amides there is significant overlap between the lone pair of the amide nitrogen and the carbonyl pi bond system. The C-O bond now has less double bond character and therefore it takes less energy to stretch. In contrast chlorine (or any halogen) is not basic and so does not participate in this kind of resonance through the pi system. In acid chlorides, the chlorine is removing electron density from the carbon by induction through the pi system; there is more C=O double bond character than in an amide and it takes more energy to stretch the carbonyl bond (leading to a higher IR absorption).
Electron donation from the nitrogen means that the lone pair on the amide nitrogen is not as available for donation to acids. This means that amides are not basic, or rather, nowhere near as basic as amines in which the lone pair is freely available for donation to an acid. This has important ramifications in biological systems. In polypeptides, which are amino acids linked by amide functional groups (circled below), amines are protonated at physiological pH (they exist as RNH3+)[8]. It is an interesting thought experiment to predict how proteins and peptides would behave if amide nitrogens were more basic. 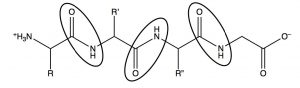 While there are relatively few naturally occurring acidic or basic side chains in polypeptides (from amino acids such as glutamic acid or lysine), every peptide is linked by innumerable amide bonds between the individual amino acids. If all these amide nitrogens were protonated, the peptides and proteins would take up very different structures (since they would have a large positive charge which would, in the absence of counter ions, repel other parts of the molecule).
While there are relatively few naturally occurring acidic or basic side chains in polypeptides (from amino acids such as glutamic acid or lysine), every peptide is linked by innumerable amide bonds between the individual amino acids. If all these amide nitrogens were protonated, the peptides and proteins would take up very different structures (since they would have a large positive charge which would, in the absence of counter ions, repel other parts of the molecule).
Relative reactivities of carboxylic acids and derivatives
The evidence from IR spectroscopy can help us predict the relative reactivities of carbonyls. For example: IR spectroscopy evidence tells us that the amide functional group is stabilized and the nitrogen lone pair is conjugated to the carbonyl group, whereas the partially positive charge at the acid chloride carbonyl carbon is increased (because of induction) compared to the amide. Therefore, a plausible prediction is that the acid chloride is more reactive than the amide and, as we shall see shortly, this is true. In general, the order of reactivity parallels the absorption frequency of the carbonyl group, acid chlorides are more reactive than anhydrides, esters and carboxylic acids are fairly similar in their reactivity (except with bases), and amides are the least reactive. In general, aldehydes and ketones are more reactive than all carboxylic acid derivatives except acid chlorides.
Reactions at the carbonyl group of acid derivatives with irreversible nucleophiles
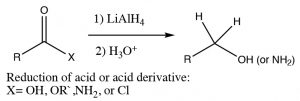 Just as we saw with aldehydes and ketones, we can reduce a carbonyl group by the addition of a hydride ion. Typically, Lithium Aluminum Hydride (rather than sodium borohydride) is used in such a reaction because acid derivatives (amides, esters, anhydrides, chlorides) are usually not reactive enough to react with sodium borohydride. With LiAlH4, the acid or acid derivative is reduced all the way down to the primary alcohol or amine. With acids or esters, the reaction does not stop at the aldehyde step since aldehydes are generally more reactive and the reducing reagent will preferentially reduce any aldehyde as it is formed.
Just as we saw with aldehydes and ketones, we can reduce a carbonyl group by the addition of a hydride ion. Typically, Lithium Aluminum Hydride (rather than sodium borohydride) is used in such a reaction because acid derivatives (amides, esters, anhydrides, chlorides) are usually not reactive enough to react with sodium borohydride. With LiAlH4, the acid or acid derivative is reduced all the way down to the primary alcohol or amine. With acids or esters, the reaction does not stop at the aldehyde step since aldehydes are generally more reactive and the reducing reagent will preferentially reduce any aldehyde as it is formed.
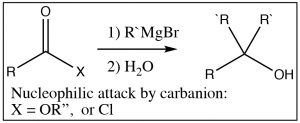 The situation is a different when acid derivatives are reacted with highly reactive carbanions such as Grignard reagents or alkyl lithium reagents. In this case, any derivative that has acidic protons (the carboxylic acid itself or most amides) will simply deprotonate and the reaction will go no further. With esters or acid chlorides, however, two equivalents of the carbanion add to the carbonyl. The reaction goes through the intermediate step of forming a ketone, but just like the LiAlH4 reduction, since ketones are more reactive than acid derivatives, the ketone will undergo nucleophilic attack as they are formed, resulting in the alcohol.
The situation is a different when acid derivatives are reacted with highly reactive carbanions such as Grignard reagents or alkyl lithium reagents. In this case, any derivative that has acidic protons (the carboxylic acid itself or most amides) will simply deprotonate and the reaction will go no further. With esters or acid chlorides, however, two equivalents of the carbanion add to the carbonyl. The reaction goes through the intermediate step of forming a ketone, but just like the LiAlH4 reduction, since ketones are more reactive than acid derivatives, the ketone will undergo nucleophilic attack as they are formed, resulting in the alcohol.
Nucleophilic addition and elimination reactions of acids and derivatives
Just as with aldehydes and ketones, the reaction of acids and derivatives with oxygen and nitrogen nucleophiles is somewhat more complex: at each step, there is the potential for reversal. The formation and decomposition of the tetrahedral intermediate again plays a central role in the outcome of the reaction, and it is possible to use the knowledge of structure and properties to predict how the reaction will proceed. Furthermore, with a knowledge of how concentrations of reactants and products affect equilibrium positions, we can control the outcome of reactions using Le Châtelier’s Principle.
First, we will consider the reaction of a carboxylic acid with an alcohol leading to the formation of an ester. 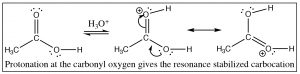 The reaction is typically performed with an acid catalyst and given that information, you should be able to write the mechanism. The first step is protonation; while there are two potentially basic oxygens, protonation tends to occur preferentially on the carbonyl oxygen (not the OH) because the resulting cation can be resonance-stabilized. As with aldehydes and ketones, protonation activates the carbonyl and the next step is attack by the nucleophile—in this case an alcohol. In the reaction scheme below, ethanol reacts with acetic acid to give ethyl acetate.
The reaction is typically performed with an acid catalyst and given that information, you should be able to write the mechanism. The first step is protonation; while there are two potentially basic oxygens, protonation tends to occur preferentially on the carbonyl oxygen (not the OH) because the resulting cation can be resonance-stabilized. As with aldehydes and ketones, protonation activates the carbonyl and the next step is attack by the nucleophile—in this case an alcohol. In the reaction scheme below, ethanol reacts with acetic acid to give ethyl acetate.
The crucial part of this mechanism is the series of tetrahedral intermediates that are interconverted by protonating and deprotonating the three different oxygens. Since all these groups are similar (OH or OR) the probability of each of these groups leaving is more or less the same once they are protonated. Just as with aldehydes and ketones, the system will become more stable (with stronger bonds) if the carbonyl group reforms by the elimination of one of the groups attached to the carbon. Again, we can shift the equilibrium for this reaction by manipulating the reaction conditions. Typically, esterifications are carried out using the alcohol as solvent (so it is in large excess), and the water produced is removed as it is formed.
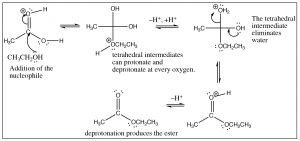
As you might expect, this reaction is entirely reversible, and the reverse reaction is typically carried out in aqueous solution with either acid or base catalysis. In fact, this reaction is the basis of saponification (soap making); in which long-chained fatty acid esters of glycerol (triglycerides) are hydrolyzed in an aqueous solution with a base catalyst.
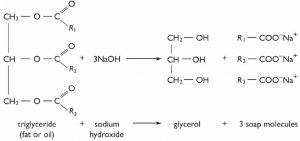
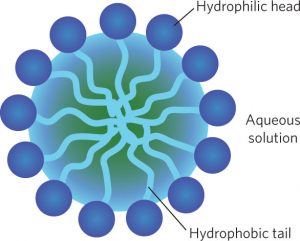
The triglyceride (fat or oil) is insoluble in water, while the sodium salt of the long-chain fatty acid (soap) is soluble. The soap molecules aggregate to form spherical micelles in which the polar head groups lie on the outside and the non-polar tails are inside[9].
Interconversion of acids and derivatives: predicting outcomes.
As we have just seen, esters can be made from carboxylic acids and vice versa: we can control the outcome of the reaction by using Le Châtelier’s principle. This is because the tetrahedral intermediate contains only oxygen leaving groups and the reactants and products are of similar stability. However, if we react a derivative such as an acid chloride with an oxygen nucleophile, we see that the resulting reaction tends not to be reversible. It is possible to go from the acid chloride to the carboxylic acid (with H2O nucleophile), to the ester (with an alcohol nucleophile), or to the amide (with an amine nucleophile). The reverse reaction is not feasible because 1) the tetrahedral intermediate, for either the forward or the reverse reaction, now has different leaving groups as shown below and 2) the acid chloride is highly reactive (it is destabilized by inductive withdrawal) and the reaction is unlikely to reverse under typical reaction conditions.
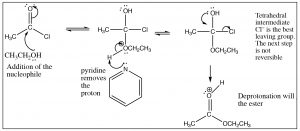
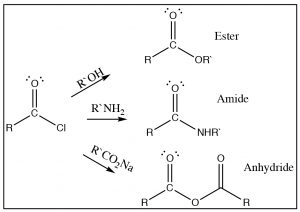 The tetrahedral intermediate reacts to produce the carbonyl by expelling chloride, which is the best leaving group. It is, therefore, difficult to get this reaction to reverse. In fact, the acid chloride can be used to produce all the other acid derivatives, and carboxylic acids. Reaction of acid chlorides with amines will produce amides and, with a carboxylate anion, will produce acid anhydrides as shown. In each case chloride is the best leaving group when the tetrahedral intermediate collapses, and the more stable product is formed.
The tetrahedral intermediate reacts to produce the carbonyl by expelling chloride, which is the best leaving group. It is, therefore, difficult to get this reaction to reverse. In fact, the acid chloride can be used to produce all the other acid derivatives, and carboxylic acids. Reaction of acid chlorides with amines will produce amides and, with a carboxylate anion, will produce acid anhydrides as shown. In each case chloride is the best leaving group when the tetrahedral intermediate collapses, and the more stable product is formed.
You may now be asking yourself if acid chlorides are such good reactants, how can we make them in the first place? We cannot use chloride ion to do a nucleophilic addition/elimination on any other acid derivative, so how can we get around that problem? The answer is to introduce an even better leaving group into the molecule. One example is the reaction of carboxylic acids with thionyl chloride (SOCl2). The sulfur in thionyl chloride is highly susceptible to nucleophilic attack—much more so than the carbonyl—because of all the electronegative groups attached to it. The first step is attack by the carboxylic acid oxygen on the SOCl2, as shown below.
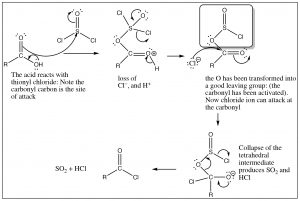
The intermediate that is formed has an excellent leaving group (in the shadowed box). In effect, we have activated the carbonyl and made it more reactive. Attack of chloride ion can now proceed and the reaction will move forward because we have a better leaving group than chloride. The tetrahedral intermediate now collapses and, at the same time, the leaving group decomposes to give SO2 and hydrogen chloride[10], both of which are gases that are expelled from the reaction mixture which drives the reaction towards products (Le Chatelier’s principle).
In this reaction, we have seen a very important and powerful idea: we have been able to drive a reaction forward to produce a thermodynamically unfavorable product by producing a highly reactive intermediate. We encountered simpler examples earlier: for example, protonating an alcohol or preparing a tosyl derivative to transform OH into a good leaving group. In this case, the reaction is messy and toxic (it’s no fun at all to do this reaction in a laboratory), but biological systems use this strategy to bring about thermodynamically unfavorable reactions. As we will discuss later in the course, substrate reactants can be activated by this same strategy of making an OH into a good leaving group that then leads to the formation of a product that could not be produced under normal circumstances. For example, the formation of sucrose from glucose and fructose proceeds using such a (enzyme-catalyzed) strategy. In this case, activation involves the activation of an OH group by coupling with ATP.
Preparations of carboxylic acids.
All the derivatives of carboxylic acids can be produced from the acid form, although it may occur via the acid chloride. 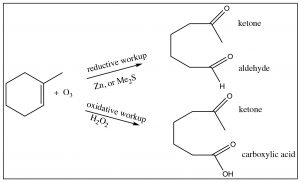 We have also seen several reactions in which carboxylic acids can be produced by alternative mechanisms: for example, via the oxidation of primary alcohols or aldehydes. It turns out that we can also produce acids by ozonolysis (reaction with ozone O3) if one or both of the alkene carbons are bonded to a hydrogen. When we looked at such reactions previously, they were accompanied by a reductive workup (either Zn, or dimethyl sulfide) and arrested at the aldehyde level. If instead we use an oxidative workup with hydrogen peroxide, the oxidation goes all the way through to the carboxylic acid.
We have also seen several reactions in which carboxylic acids can be produced by alternative mechanisms: for example, via the oxidation of primary alcohols or aldehydes. It turns out that we can also produce acids by ozonolysis (reaction with ozone O3) if one or both of the alkene carbons are bonded to a hydrogen. When we looked at such reactions previously, they were accompanied by a reductive workup (either Zn, or dimethyl sulfide) and arrested at the aldehyde level. If instead we use an oxidative workup with hydrogen peroxide, the oxidation goes all the way through to the carboxylic acid.
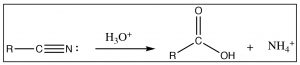 Another reaction that is often used to produce carboxylic acids is the hydrolysis of nitriles (RCN). The nitrile carbon is at the same oxidation state as a carboxylic acid, and when treated with aqueous acid (or base), it undergoes a hydrolysis reaction in just the same way as we have seen on numerous occasions (try it—you will see!).
Another reaction that is often used to produce carboxylic acids is the hydrolysis of nitriles (RCN). The nitrile carbon is at the same oxidation state as a carboxylic acid, and when treated with aqueous acid (or base), it undergoes a hydrolysis reaction in just the same way as we have seen on numerous occasions (try it—you will see!).
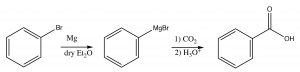
Finally, a different approach to producing carboxylic acids occurs when we react a Grignard reagent (or alkyl lithium) with carbon dioxide. You may recall from general chemistry that CO2 is a linear molecule and has no overall molecular dipole. However, each C=O bond in the molecule is polarized, leading to a partial positive charge on the central carbon that makes it susceptible to nucleophilic attack. Therefore, when reacted with a Grignard reagent, the end result is the formation of a new C-C bond with the CO2 becoming a carboxyl group.
The Wittig Reaction
This reaction allows us to synthesize alkenes by adding the two carbons of the alkene double bond together. This is a very powerful synthetic technique that allows us to construct larger molecules from smaller ones. It is much more efficient to add two molecules together, than to try and synthesize a large molecule one carbon-carbon bond at a time.
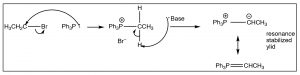
The Wittig reaction starts by preparation of a reagent that involves addition of an alkyl halide to a phosphine (the phosphorus analog of an amine), such as triphenylphosphine (Ph3P). This reaction is directly analogous to the alkylation of amines that we have seen previously to produce the phosphonium salt as shown. However, the reaction can go further in the presence of a base to produce the stabilized carbanion by deprotonation of the carbon next to the phosphorus as shown[11].[12]
The Wittig reagent can then react with an aldehyde or ketone as shown. The first step, as we might imagine, involves nucleophilic attack at the carbonyl, but since the carbanion is also attached to the phosphorus, another avenue of reaction is now available. The tetrahedral intermediate undergoes further reaction in which the oxygen bonds to the phosphorus and then, via a cyclic rearrangement of electrons, the four-membered ring rearranges to form the alkene and triphenylphosphine oxide. 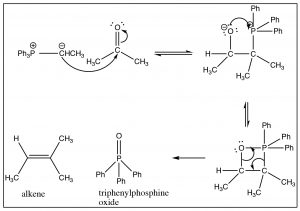 In fact, it is the formation of the very strong P=O double bond that makes this reaction essentially irreversible, and drives the reaction towards products.
In fact, it is the formation of the very strong P=O double bond that makes this reaction essentially irreversible, and drives the reaction towards products.
The Wittig reaction can be used to prepare many different alkenes. In general, if there is a possibility of E/Z isomerism, the most stable alkene will be produced (for example a trans isomer rather than a cis isomer). But, just as we have seen many times, it is possible to manipulate the reactants and reaction conditions to produce the desired product—a subject for a more advanced treatment of organic chemistry[13].
Synthesis
Up to now, we have focused mostly on the reactivity of organic molecules, particularly from the standpoint of being able to predict reactivity from the structure of a particular compound. We have also discussed how spectroscopic techniques allow us to determine molecular structure. However, we have not really discussed another of the major areas of organic chemistry, that is: the design and synthesis of molecules, particularly those that may be expected to have biological activity (drugs of various sorts). Now that we have a fairly large repertoire of reactions to choose from, let us take a closer look at the kinds of decision-making that goes into designing molecular structures.
Typically, molecular synthesis involves a target structure, which may be an actual molecule, or it could be a substance that has particular properties: for example, the active site of an enzyme or a regulatory domain of a protein. To achieve specificity, the molecule to be synthesized must fit into the surface of the protein and influence its structure or catalytic activity. The better the fit, the more specific (higher affinity, fewer non-target interactions) interactions the drug will make. We will begin by thinking about how to go about the synthesis of a given molecule. Molecular synthesis is both an art and a science: it requires that you have at your fingertips a good collection of reactions you have organized in such a way as to make them accessible to you, but it also requires creativity and imagination. There is always more than one way to design a synthesis and, in reality, there are many setbacks and path changes since reactions may not go as planned, so alternative routes have to be considered. In this section, we will look at strategies that you might employ to design a synthesis of a target molecule.
Retrosynthetic analysis
Retrosynthetic analysis is exactly what it sounds like: you begin with the target and move backwards one step at a time to identify what reactants and reagents could have produced the products. 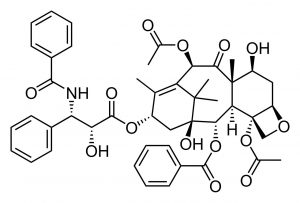 This is often the situation when you are dealing with a natural product that was originally isolated based on its biological activity. A classic example is the molecule Paclitaxel, which was isolated from the Pacific yew tree, Taxus bervifolia, based on its anti-cancer properties.[14]
This is often the situation when you are dealing with a natural product that was originally isolated based on its biological activity. A classic example is the molecule Paclitaxel, which was isolated from the Pacific yew tree, Taxus bervifolia, based on its anti-cancer properties.[14]
The process of molecular synthesis can be broken down into a number of steps, but as we will see, depending on the nature of the task, we do not always approach synthesis in a linear manner.
Step 1: Identify the number of carbons in the target molecule and determine whether you will need (at some point in the synthesis) to make new carbon-carbon bonds.
Step 2: Identify what functional groups are present in the molecule. Functional groups are where the reactivity of any molecule lies and they give you a place to begin because you now know ways to produce that functional group.
Step 3: Identify which bonds are going to be made during the reaction in which the product is produced.
Step 4: This will allow you to work backwards to identify what the precursor is and (using your knowledge of functional group transformations) decide what reaction will product the product.
Step 5: Repeat until you reach a recognizable target material.
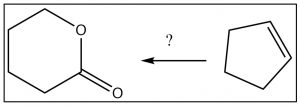 Let us approach the process with a (significantly simpler) target.[15] Let us design a synthesis of the lactone (cyclic ester) from cyclopentene. We begin by noting that both the starting material and the product have five carbons, which means we do not need to consider any carbon-carbon bond formation reactions. However, the route from cyclopentene to the lactone is not obvious, so let us go back one step at a time as outlined below.
Let us approach the process with a (significantly simpler) target.[15] Let us design a synthesis of the lactone (cyclic ester) from cyclopentene. We begin by noting that both the starting material and the product have five carbons, which means we do not need to consider any carbon-carbon bond formation reactions. However, the route from cyclopentene to the lactone is not obvious, so let us go back one step at a time as outlined below.

So, we have the immediate precursor to the lactone, but how do we get that from cyclopentene? While you could continue moving backwards through the steps, the knowledge that you don’t have to construct the carbon skeleton makes a difference in the way that you might approach the problem. For example, if you look at the starting material, it is clear that there is no five-membered ring in the product and no alkene. Not only that, but at each end of the carbon chain is at least one oxygen. Is there a reaction that will allow us to open up that ring and, at the same time, introduce oxygens at each carbon? Yes! Recall that alkenes can be cleaved by ozonolysis (under oxidizing conditions), to give the dicarboxylic acid. If one of those carboxylate groups can be reduced to the alcohol, something that might well be be difficult to do in practice, the ring would cyclize spontaneously to give the lactone as shown below.
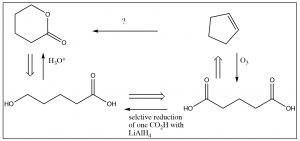
Note that, in this synthesis, we mixed up both retro and forward synthetic analysis to produce an overall synthetic pathway. This is where some of the art and imagination come into play. When you are trying to design a synthesis, it is a good idea to start from the product and work backwards—but at some point you may have to also work forward. Every synthesis is different and, as we noted before, there are often many synthetic routes that can be designed for any single target molecule.
- IUPAC is the International Union of Pure and Applied Chemists. This body is responsible (among other things) for setting the rules about systematic nomenclature of chemical substances. ↵
- The preparation of aldehydes and ketones has been discussed earlier, including reactions in which alkenes are cleaved (broken apart) by oxidation with ozone (ozonolysis) by addition of water across triple bonds (Chapter 5) and the oxidation of alcohols (Chapter 6). ↵
- Why do you think that pyruvate and lactate are present in the form of their conjugate bases? ↵
- Sometimes this grouping is called a ketal (when the starting C=O is a ketone), but general “acetal” and “hemiacetal” can refer to either an aldehyde or a ketone. ↵
- Remember, Le Chateliers Principle is just a rule of thumb—it tells us what happens but not why. Adding more reactants increases the rate of the forward reaction, removing products decreases the rate of the reverse reaction. ↵
- However, as we will see later, carbonyl compounds are often acidic; the alpha carbon can be deprotonated; more on that later. ↵
- See chapter 9 of CLUE general chemistry text for more information on the Henderson Hasselbalch equation and its uses. ↵
- The pKa of protonated amines (RNH3+) is about 10. Using the Henderson Hasselbalch equation, we see that the ratio of [RNH2]/[RNH3+] is about 0.001—that is, there is 1000 times more protonated than unprotonated amine. ↵
- For a more in-depth discussion of this phenomenon, including the entropic and enthalpic contributions to micelle formation, see the CLUE Chapter 6. ↵
- Both HCl and SO2 are highly toxic, requiring special precautions—another one not to try at home. ↵
- Note that this stabilization uses d orbitals on the phosphorus; this reaction could not happen with an amine. ↵
- Species such as this carbanion are called ylides, because they can be written as containing both negative and positive charges on adjacent atoms (in contrast to Zwitterions: forms of amino acids at different pH’s that also have both positive and negative charges on them—just not on adjacent atoms). ↵
- See http://www.organic-chemistry.org/namedreactions/wittig-reaction.shtm for example. ↵
- It functions (In at least one way, by binding to the protein tubulin: the structural basis of the cytoplasmic microtubules found in eukaryotic cells. When bound, Paclitaxel acts to make microtubules more stable (i.e. less likely to depolymerize). Since microtubule function depends on dynamic assembly and disassembly, this has effects on cell behavior, specifically microtubule-based cell division ↵
- The total synthesis of taxol is described here: http://www.nature.com/nature/journal/v367/n6464/abs/367630a0.html ↵
