
Chapter 6: Alcohols and an introduction to thiols, amines, ethers & sulfides
In this chapter, we are going to take a closer look at the families of compounds that have carbon linked through a single covalent bond to an O, N, or S. These are known as alcohols (R-OH), amines (R-NH2, RR’-NH, RR’R”-N), thiols (R-SH), ethers (R-OR’), and sulfides (R-SR’). We group these compounds together based on the predictable similarities and differences in their chemical and physical properties, specifically the fact that each of these functional groups has a relatively electronegative element (O, N or S) attached by a single bond to carbon and each has available lone electron pairs that can be donated to H+ or other electrophiles. The result is that alcohols, thiols, and amines (primary and secondary) all have relatively acidic hydrogens, which influences their chemical reactivities, and all show nucleophilic properties.
Table 6.1 Examples of Functional groups, their names and approximate pKa‘s
| Functional Group | Example | Name | pKa |
| Alcohol | 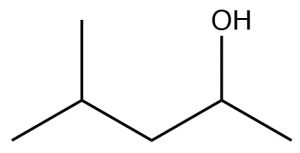 |
Remove -ane, add -ol.
4-methylpentan-2-ol |
(approximate)
~15-16 |
| Alcohol | 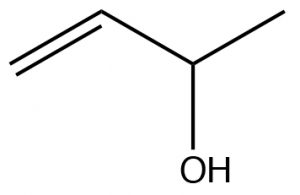 |
Alcohols take precedence over
alkenes, But-3-en-2-ol |
|
| Thiol | 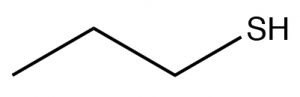 |
Longest chain, add -thiol
Propane-1-thiol |
~10 |
| Primary amine | 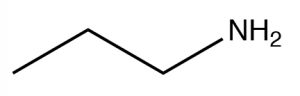 |
Longest chain, remove -e, add
-amine propanamine or propyl amine |
~33 |
| Secondary amine | 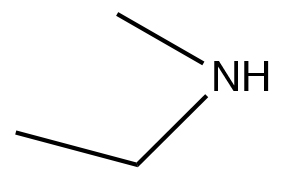 |
N-methylethanamine | ~33 |
| Tertiary amine | 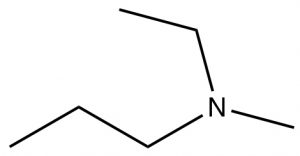 |
N-ethyl-N-methylpropanamine | N/A |
| Ether | 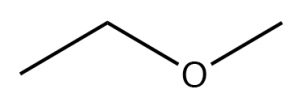 |
Methoxyethane
Ethyl methyl ether |
N/A |
| Sulfide | 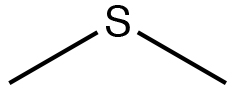 |
Dimethylsulfane
Dimethyl sulfide |
N/A |
We will concentrate our discussion on oxygenated compounds, but we will note reactivities across the various groups to illustrate their similarities (and differences).
(Brønsted) Acidity of alcohols, thiols, and amines
Recall that there are several factors that can influence Brønsted acidity. These include: the strength of the bond between the R (C) and the O, S, or N (denoted by “Y” below) and H; the polarity of the bond; and the stability of the resulting anion (“Y–“).
R–Y–H + B– [latex]\rightarrow[/latex] BH + R–Y–
Simple alcohols have acidities that are about the same as water, with a pKa of around 15-16 (remember the pKa of water is 14). In comparison, amines are much less acidic, with pKa‘s around 33-36. We understand this difference based on the fact that O is more electronegative than N, therefore the O–H bond is more polarized than the N–H bond—making the partial positive charge on the H larger in O–H than in N–H bonds. In polar solvents, this means that the H bonded to an O is better solvated and the proton is easier to remove than an H bonded to an N. Similarly, the resulting negative charge on the anion is more stable on O than on N because the effective nuclear charge on O is greater (which is the cause of its greater electronegativity, compared with N). In contrast, thiols are more acidic than alcohols because the S–H bond is weaker—the size of S and H orbitals results in smaller overlap and therefore weaker bonds (just like HBr is more acidic than HCl) and the resulting anion is more stable because the larger size of S results in the negative charge being distributed over a larger volume.
To deprotonate a simple alcohol, with a pKa of around 15, requires a base that forms a bond with H that is than the bond that would be formed with the resultant alkoxide (RO–)—otherwise the acid-base reaction would reverse. For this reason, we cannot use a base like sodium hydroxide, because its conjugate acid H2O has a pKa of around 14. Therefore, although some alcohol deprotonation would occur in water, the equilibrium position would be somewhere in the middle, so that both –OH and RO– would be present in the reaction mixture. To completely deprotonate an alcohol with a pKa of around 15, we would need to use a stronger base, such as sodium hydride[1] (NaH), or sodium amide[2] (NaNH2).
![]()
Note that the reaction must take place in an aprotic solvent; in this case diethyl ether is used (otherwise the NaH would react with the solvent as well as the alcohol). Another common method to deprotonate alcohols is through a redox reaction, using a group one metal—usually sodium.
![]()
Some alcohols are much more acidic; for example –OH groups attached to an aromatic ring (which are called phenols) typically have pKa‘s around 10. This difference in pKa must lie with the nature of the conjugate base (the anion), since the same O–H bond breaks during the proton transfer. In this case, the conjugate base is stabilized by delocalizing the electrons to the aromatic ring by resonance. Recall that the more delocalized a charge (in this case the electrons), the more stable the resultant ion is.
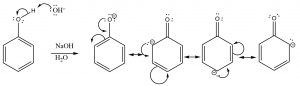 Phenols are more acidic than typical alcohols because the conjugate base is stabilized by resonance
Phenols are more acidic than typical alcohols because the conjugate base is stabilized by resonance
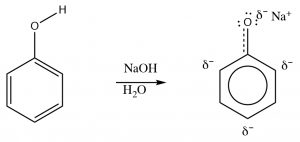 Phenols can be deprotonated by NaOH because the phenolate anion is more stable than hydroxide. Therefore, phenols are soluble in aqueous solutions of sodium hydroxide. This also provides a way of separating phenols from other (non-acidic) organic substances, since the phenol can be regenerated simply by adding acid. One practical way that this phenomenon can be used is to remove the highly allergenic substances secreted by poison ivy (or oak or sumac) plants.
Phenols can be deprotonated by NaOH because the phenolate anion is more stable than hydroxide. Therefore, phenols are soluble in aqueous solutions of sodium hydroxide. This also provides a way of separating phenols from other (non-acidic) organic substances, since the phenol can be regenerated simply by adding acid. One practical way that this phenomenon can be used is to remove the highly allergenic substances secreted by poison ivy (or oak or sumac) plants. 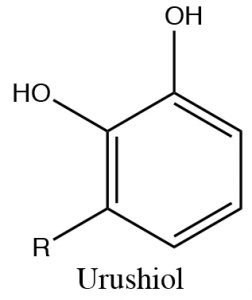 The major allergen belongs to a family of di-phenols called urushiol. The R can by any of a number of long chained hydrocarbons. Washing the affected part with a basic solution (soap for example) will help solubilize the urushiol and remove it from the skin.
The major allergen belongs to a family of di-phenols called urushiol. The R can by any of a number of long chained hydrocarbons. Washing the affected part with a basic solution (soap for example) will help solubilize the urushiol and remove it from the skin.
Alcohol acidity can also be increased by inductive electron withdrawal (due to the presence of electronegative atoms linked through sigma bonds) just as we discussed earlier in the case of carboxylic acids: for example CF3OH is more acidic than CH3OH.
We might also predict the effects relative to acidities of amines and thiols in terms of resonance and inductive stabilization, but, in fact, most of their chemistry is not associated with acidity and we will not dwell on this idea here.
Nucleophilicity of ROH, RSH, and RNH2
Earlier (Chapters 1 and 4), we discussed (at great length) that all three functional groups (–OH, –NH, and –SH) are nucleophilic: that is, they will react at the carbon center that is electron-deficient. For functional groups that contain nucleophilic centers from the same row of the periodic table, the trends in nucleophilicity parallel Bronsted basicity: amines are more nucleophilic (and basic) than alcohols. However, in functional groups that contain nucleophilic centers from the same group of the periodic table (nucleophilicity increases down the group, while basicity decreases), thiols are more nucleophilic than alcohols. Both amines and thiols are very nucleophilic. All three groups participate in nucleophilic substitutions as discussed in Chapters 1 and 4.
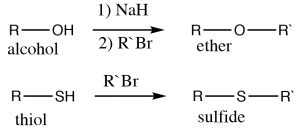 Examples of these kinds of nucleophilic substitutions are the reactions of alcohols, thiols, and amines with alkyl halides to give the corresponding ethers, sulfides, and (secondary, tertiary or quaternary) amines. Alcohols are not as nucleophilic as thiols and amines, and therefore typically the corresponding alkoxide must be used (because it is more reactive), for the synthesis of ethers.
Examples of these kinds of nucleophilic substitutions are the reactions of alcohols, thiols, and amines with alkyl halides to give the corresponding ethers, sulfides, and (secondary, tertiary or quaternary) amines. Alcohols are not as nucleophilic as thiols and amines, and therefore typically the corresponding alkoxide must be used (because it is more reactive), for the synthesis of ethers.
In the case of amines, the nitrogen can react several times with the electrophile (alkyl halide), and in practice it is difficult to stop the reaction at any intermediate step (in the laboratory).
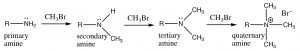
Amines typically react with electrophiles to give poly-alkylated amines
O, S, and N as leaving groups:
Recall that a good leaving group should be able to accept (in a stable form) the pair of electrons from the bond that breaks. Typically, good leaving groups are weak bases. For this reason, hydroxide (–OH) and amide (–NH2) are unlikely to be produced during a nucleophilic substitution reaction. However, as noted earlier, alcohols can be converted into good leaving groups by protonation, which results in H2O as the leaving group.
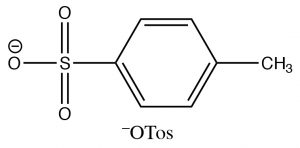 Alcohols can also be modified (or derivatized) to produce better leaving groups. This is particularly useful when we need to carry out a reaction that is sensitive to acidic conditions when the method we have used earlier (protonation of the OH) cannot be used. The most common derivative used to make the OH group into a good leaving group is the Tosyl group (para-toluenesulphonate). It can be formed by reacting an alcohol with p-toluenesulfonylchloride (TosCl) in the presence of a base (such as pyridine) that acts to remove the HCl that is produced).
Alcohols can also be modified (or derivatized) to produce better leaving groups. This is particularly useful when we need to carry out a reaction that is sensitive to acidic conditions when the method we have used earlier (protonation of the OH) cannot be used. The most common derivative used to make the OH group into a good leaving group is the Tosyl group (para-toluenesulphonate). It can be formed by reacting an alcohol with p-toluenesulfonylchloride (TosCl) in the presence of a base (such as pyridine) that acts to remove the HCl that is produced).
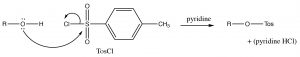
We can consider the derivatization reaction as mechanistically similar to other nucleophilic substitutions we have considered, except that it takes place at an S instead of a C.
The resulting OTos group is a very good leaving group, making the molecule reactive to nucleophilic substitution reactions. In effect, we have changed the leaving group from –OH, which is a relatively strong base, to –OTos which is a very weak base—it is the organic equivalent of sulfate, the conjugate base of sulfuric acid. The negative charge on –OTos becomes delocalized to the other oxygens bound to the S, thereby stabilizing the base.
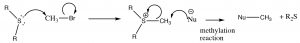
In a similar manner, sulfides can be transformed into leaving groups, most commonly through the methylation of the sulfide, which produces a powerful reagent that can be used to methylate other species.
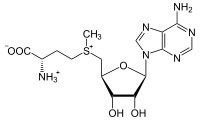 In biological systems, a common methylating agent, S–adenosylmethioninine (SAM [latex]\rightarrow[/latex] ), uses this mechanism.
In biological systems, a common methylating agent, S–adenosylmethioninine (SAM [latex]\rightarrow[/latex] ), uses this mechanism.
Oxidation of Alcohols
Before we discuss oxidation of alcohols, it should be clear what we mean by the “oxidation” and “reduction” of carbon compounds. Recall that in earlier discussions we used the term reduction to mean the addition of hydrogen and oxidation to mean the addition of oxygen, rather than calculating changes in oxidation numbers (decrease for reduction, increase for oxidation). The reason is because oxidation numbers in organic compounds can be hard to calculate and apply[3]. In this section, we consider how alcohols can be oxidized to give aldehydes, ketones, or carboxylic acids. In general, we consider a carbon compound to be oxidized when the number of bonds between the C and electronegative atoms (often, but not always, O) is increased.
For example, a primary alcohol can be oxidized (which we will denote by O for the time being) to an aldehyde; depending upon the reagent used, the reaction can proceed through a second step to produce the corresponding carboxylic acid. At each step, the oxidation level of the carbon is increasing.
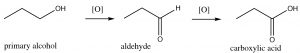
Starting with a secondary alcohol, the product of an oxidation reaction is the corresponding ketone, but tertiary alcohols do not give useful products and may simply lead to degradation (C–C bond breaking). Generally, it is not possible to oxidize a secondary carbon beyond the ketone level without breaking carbon-carbon bonds, and similarly, tertiary alcohols cannot be oxidized under normal circumstances.
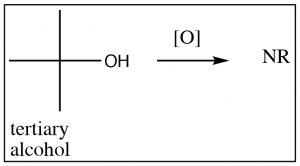
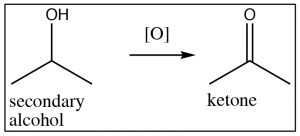
Typical oxidizing reagents include transition metals in high-oxidation states (that is able to accept [bond to] O atom).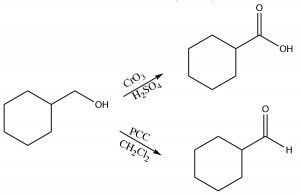 For example, chromium (VI) in the form of chromium trioxide (CrO3) or sodium dichromate (Na2Cr2O7), when in concentrated H2SO4, are both powerful oxidizing agents and both will oxidize a primary alcohol through both steps, that is, all the way to the carboxylic acid form. Pyridinium chlorochromate (PCC [latex]\rightarrow[/latex]) is a milder oxidizing agent that will oxidize primary alcohols, only through the first step, to produce an aldehyde.
For example, chromium (VI) in the form of chromium trioxide (CrO3) or sodium dichromate (Na2Cr2O7), when in concentrated H2SO4, are both powerful oxidizing agents and both will oxidize a primary alcohol through both steps, that is, all the way to the carboxylic acid form. Pyridinium chlorochromate (PCC [latex]\rightarrow[/latex]) is a milder oxidizing agent that will oxidize primary alcohols, only through the first step, to produce an aldehyde.
The general mechanism of oxidation is shown below, note electrons leave the alcohol and end up on the Cr, reducing its oxidation state from 6 to 4, and the alcohol carbon ends up oxidized.
Primary alcohols can be selectively oxidized to aldehydes with PCC
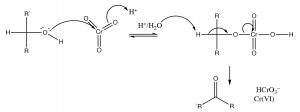
 One problematic aspect of such oxidizing reagents is that they contain highly toxic and carcinogenic Cr (VI) in one form or another. Such materials oxidize a range of biomolecules such as vitamin C (ascorbic acid) and some thiols (such as the amino acid cysteine). Reduced chromium also reacts with nucleic acids and can lead to mutations, which can lead to cell death and/or cancer. To avoid using such toxic chemicals, there has been increasing in what has come to be known as green chemistry[4]. One of the tenets of green chemistry is to minimize the use of toxic reagents (such as chromium compounds).[5]
One problematic aspect of such oxidizing reagents is that they contain highly toxic and carcinogenic Cr (VI) in one form or another. Such materials oxidize a range of biomolecules such as vitamin C (ascorbic acid) and some thiols (such as the amino acid cysteine). Reduced chromium also reacts with nucleic acids and can lead to mutations, which can lead to cell death and/or cancer. To avoid using such toxic chemicals, there has been increasing in what has come to be known as green chemistry[4]. One of the tenets of green chemistry is to minimize the use of toxic reagents (such as chromium compounds).[5]
Oxidation of Thiols
In alcohols, oxidation generally occurs at the carbon bonded to oxygen. In contrast, with thiols the oxidation site is often at the sulfur. For example, many oxidizing agents (even molecular oxygen in air) oxidize thiols to disulfides. The reverse reaction is also readily accomplished using a reducing agent such as Zn/HCl. The disulfide bond is relatively weak, that is, requires less energy to break (about half the strength of a typical C-C or C-H bond).[6]
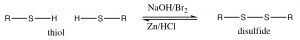
In fact, the amino acids cysteine and diamino acid cystine are readily interconverted in biological systems (usually through the NADH/NAD oxidation/reduction system; see below). These disulfide crosslinks between cysteine moieties in polypeptides and proteins often serve to stabilize the 3D structure of proteins. 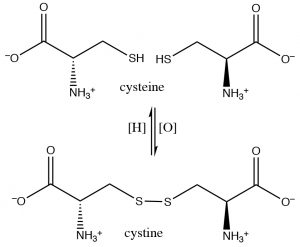
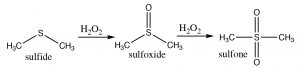
Sulfides (R-S-R) are also susceptible to oxidation, which can lead to the formation of a sulfoxide, which can be further oxidized to form a sulfone.
Preparation of alcohols
We have already seen several methods by which alcohols can be produced, mostly in Chapter 5. For example, the addition of water across a double bond, either through acid catalysis (Markovnikov addition) or by hydroboration/oxidation (Anti-Markovnikov addition), produces alcohols. We have also seen, under certain conditions, that alcohols can be produced by nucleophilic substitution. Both SN1 and SN2 reactions can produce alcohols, and now would be a good time to review all of these reactions (covered in Chapters 1, 3, 4 and 5).
A reaction that we have not yet encountered is the reduction of carbonyl compounds. For example, a ketone such as acetone can be reduced through a reaction with sodium borohydride (NaBH4) or lithium aluminum hydride (LiAlH4)[7]; both of these molecules can deliver hydride (H–) to the partially positive carbon of the carbonyl.
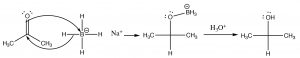
Sodium borohydride (NaBH4) is generally the reagent of choice as it is less reactive and the reaction can be carried in an open flask, whereas LiAlH4 typically must be used with solvents that do not contain water and under a dry atmosphere. The intermediate R–O–BH3 complex is destroyed by adding aqueous acid to give the final alcohol product.
Reactions where hydride is delivered to a carbonyl are similar to a reaction found in biological systems. NADH (Nicotinamide Adenine Dinucleotide Hydride) is an unstable intermediate generated through a number of metabolic processes (such as fermentation), while not as reactive as NaBH4, and (like, essentially, all biological reactions) requires a catalyst (an enzyme) to bring about the reduction of carbonyls; but the mechanism is similar. The reaction (like all reactions) is also reversible, so that the oxidized version of NADH, NAD+, can accept a hydride from an alcohol to produce a ketone. In the mechanism (with only the nicotinamide part of NADH shown) the “R” group attached to the N in the ring is actually a complex molecule consisting of an adenine moiety (a base found in nucleic acids and nucleotides), two sugar units (ribose), and two phosphate linkages. For now, let us focus on the similarities between the reduction reactions discussed above and those that take place in biological systems.
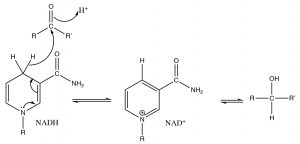
Reduction of a carbonyl by NADH by delivery of H– to the carbonyl carbon
The conversion of pyruvic acid to lactic acid during glycolysis is just such an example. By looking at simpler systems, we can understand (and model) the types of reactions that occur in organisms.
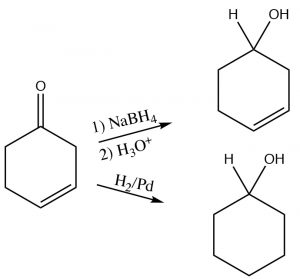 Alcohols can also be produced by direct reduction with H2(g) using a transition metal catalyst, in a way similar to the reduction of C=C, except that the hydrogens add across the C=O.
Alcohols can also be produced by direct reduction with H2(g) using a transition metal catalyst, in a way similar to the reduction of C=C, except that the hydrogens add across the C=O.
The choice of reducing agent depends on presence of other functional groups within the molecule. For example, if we wanted to reduce a carbonyl group in a molecule that also had a carbon-carbon double bond, we would not use H2/Pd as the reagent/catalyst, since it would also reduce the double bond as shown here (→).
Preparation of alcohols with Grignard reagents: Just as we can add hydride ion by a nucleophilic attack at a carbonyl, we can also add an alkyl group, which formally contains a carbanion (a negatively charged carbon). The most common way to do this is via a Grignard[8] reagent, produced by reacting an alkyl halide with magnesium metal in a dry atmosphere with a non-protic solvent such as diethyl ether (Et2O). The resulting Grignard reagent, RMgBr, is now polarized with a large partial negative charge on the carbon.
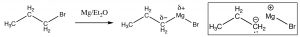
For our purposes, we can treat the Grignard as if it were a carbanion, which will react with a carbonyl group in much the same way as a hydride ion.[9]
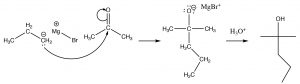
Reaction of a Grignard reagent with a ketone to give an alcohol
This reaction is applicable to a wide range of compounds that contain carbonyl groups including aldehydes and ketones, and (as we will see later) to esters and acid chlorides, but not carboxylic acids (why do you think that is?).
- NaH is synthesized by the reaction of sodium with hydrogen—a redox reaction. It is an ionic compound consisting of Na+ and H– (hydride) ions; hydride cannot be produced by deprotonating H2. ↵
- The pKa of NH3 (the conjugate acid of NH2–) is 33 ↵
- To use the oxidation number method, we must remember that H is less electronegative than C; so in CH4, the ON of carbon is –4 and each H is +1. (This is confusing since we usually consider C–H bonds as non-polar). In CO2, each O is –2 and the C is +4. Therefore, in CO2 the carbon is in a higher oxidation state than in CH4 ↵
- For more information about green chemistry see: https://www.epa.gov/greenchemistry ↵
- It is not necessary here to provide a long list of such reagents since many of them are complex, but it is important to know that there are alternatives should you ever need to oxidize an alcohol. ↵
- The formation of the analogous peroxide O–O bond (Bond Dissociation Energy 140 kJ/mol) is even less likely, this bond is even weaker than S-S (BDE 230 kJ/mol). ↵
- NaBH4 and LiAlH4 both contain a group III element (B, Al) here found in the form of the Lewis acid-base complex BH4 or AlH4. They are sources of Hydride ion, as shown above. LiAlH4 is more reactive than NaBH4. ↵
- Victor Grignard won a Nobel prize for this discovery: https://en.wikipedia.org/wiki/Victor_Grignard. ↵
- The reaction mechanism is a little more complex than this—actually occurring via one electron transfer—but the result is the same. ↵
