
Chapter 5: Alkenes and Alkynes
When a carbon is bonded to one or more electronegative atoms, it takes on a partial positive charge and it is electrophilic. Such electrophilic carbons can undergo nucleophilic substitution or elimination reactions, or both, depending upon the structures of the reacting molecules, the strength of the nucleophile, and the type of solvent in which the reaction occurs. Now, we turn to reactions that electron-rich carbon species can undergo.
Alkenes and alkynes. Both alkenes and alkynes are “unsaturated,” which means that they contain double or triple carbon-carbon bonds. The term unsaturated comes from the fact that more H atoms can be added to these molecules across the double or triple bonds. A simple alkene contains a pair of carbons linked by a double bond; this double bond consists of a sigma bond and a pi bond. 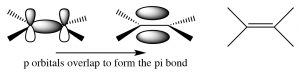 The sigma bond is formed by end-to-end overlap of sp2 hybrid orbitals, and the pi bond by side-to-side overlap of the p orbitals. A pi bond has two lobes of electron density above and below the plane of the molecule. There are a number of consequences to this arrangement: 1) the resulting region of the molecule is planar (the molecule is said to have trigonal planar geometry), 2) the electron density between the two carbons is high because there are four electrons in this region instead of two, and 3) rotation around a double bond is constrained (in contrast to rotation around a single bond). Rotation around a double bond requires breaking the overlap of the pi bond and its subsequent reformation. As with all bond-breaking phenomena, the bond-breaking step requires energy; in fact, significantly more energy than is required to bring about rotation around a single bond where no bond-breaking occurs. As we will see, these three factors have a marked effect on the behavior of alkenes.
The sigma bond is formed by end-to-end overlap of sp2 hybrid orbitals, and the pi bond by side-to-side overlap of the p orbitals. A pi bond has two lobes of electron density above and below the plane of the molecule. There are a number of consequences to this arrangement: 1) the resulting region of the molecule is planar (the molecule is said to have trigonal planar geometry), 2) the electron density between the two carbons is high because there are four electrons in this region instead of two, and 3) rotation around a double bond is constrained (in contrast to rotation around a single bond). Rotation around a double bond requires breaking the overlap of the pi bond and its subsequent reformation. As with all bond-breaking phenomena, the bond-breaking step requires energy; in fact, significantly more energy than is required to bring about rotation around a single bond where no bond-breaking occurs. As we will see, these three factors have a marked effect on the behavior of alkenes.
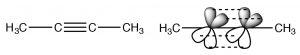 Alkynes are compounds that contain triple bonds. The triple bond consists of one sigma bond formed from end-to-end overlap of sp-hybrid orbitals and two pi bonds formed from side to side overlap. The carbons are sp-hybridized and the molecule is linear in the region of the triple bond; again rotation around a triple bond is constrained—two pi bonds must be broken for it to occur (which requires an input of energy). This bonding arrangement results in a very electron rich C-C region with the sigma bond inside what looks like a cylinder of pi electron density.
Alkynes are compounds that contain triple bonds. The triple bond consists of one sigma bond formed from end-to-end overlap of sp-hybrid orbitals and two pi bonds formed from side to side overlap. The carbons are sp-hybridized and the molecule is linear in the region of the triple bond; again rotation around a triple bond is constrained—two pi bonds must be broken for it to occur (which requires an input of energy). This bonding arrangement results in a very electron rich C-C region with the sigma bond inside what looks like a cylinder of pi electron density.
Naming Alkenes
Since alkenes have restricted rotation around the C=C group, they can exist as stereoisomers. For example, in 2-butene there is a methyl and an H bonded to each of the double-bonded carbons (carbons 2 and 3 of the molecule).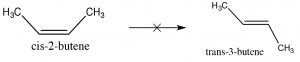 Because the C=C group is planar, the CH3 groups can be on either the same (“cis”) or opposite (“trans”) sides of the double bond (→); this cis/trans nomenclature is similar to that we used with cyclohexane rings. As the groups attached to each carbon get more complex, such nomenclature quickly becomes confusing. To cope, we turn to another established naming scheme; in this case, the Cahn-Ingold-Prelog convention we previously used with chiral centers. This involves ranking the groups linked to each double-bond carbon. If the high groups are together (same side), the name is prefixed by Z (from the German word for together: zusammen). If they are on opposite sides, they are labeled E (entgegen; away).
Because the C=C group is planar, the CH3 groups can be on either the same (“cis”) or opposite (“trans”) sides of the double bond (→); this cis/trans nomenclature is similar to that we used with cyclohexane rings. As the groups attached to each carbon get more complex, such nomenclature quickly becomes confusing. To cope, we turn to another established naming scheme; in this case, the Cahn-Ingold-Prelog convention we previously used with chiral centers. This involves ranking the groups linked to each double-bond carbon. If the high groups are together (same side), the name is prefixed by Z (from the German word for together: zusammen). If they are on opposite sides, they are labeled E (entgegen; away). 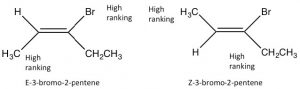 E and Z isomers are diastereoisomers: they have the same connectivity but neither can be superimposed on its mirror image. In E-3-bromo-2-pentene, the CH3 and CH2CH3 groups are closer to one another than they are in Z-3-bromo-2-pentene; the result is that they have different physical and chemical properties. These differences make it possible to separate E and Z isomers (and cis/trans since they are just a special case of E/Z) from one another.
E and Z isomers are diastereoisomers: they have the same connectivity but neither can be superimposed on its mirror image. In E-3-bromo-2-pentene, the CH3 and CH2CH3 groups are closer to one another than they are in Z-3-bromo-2-pentene; the result is that they have different physical and chemical properties. These differences make it possible to separate E and Z isomers (and cis/trans since they are just a special case of E/Z) from one another.
Stability of alkenes: Elimination reactions that produce alkenes tend to favor the most substituted alkene as the major product. The relative stabilities of various alkenes can be determined by reacting the alkene with hydrogen and determining the enthalpy change (ΔH).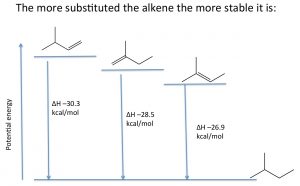
For example, shown (→), the three different alkenes produce the same product, and therefore the differences in the energy released must arise from the fact that the initial alkenes have different energies. The more alkyl groups attached to the double bond, the more stable (less reactive) the alkene is, and therefore a lower amount of energy is released. Molecular stability in alkenes is attributed to the same causes as the relative stabilities of carbocations; alkyl groups stabilize the pi bond by hyperconjugation and induction.
Reactions of Alkenes: Electrophilic Addition
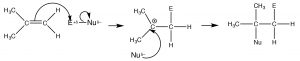
The double-bonded carbons of an alkene are electron-rich, that is, the electron density is high in the region of the double bond. Therefore, the “signature” reaction of alkenes involves initial attack on an electrophile. Instead of a substitution, alkenes undergo electrophilic addition, a reaction in which a two-component reactant adds across the double bond. The reaction begins with an electrophilic attack by the double bond onto the reactant which produces a carbocation that then undergoes nucleophilic attack. In the case of unsymmetrical alkenes (where the groups attached to the double-bonded carbons are not exactly the same), the most stable carbocation is produced. This reaction is regioselective, that is, we can predict the orientation of reactant addition across the double bond. If we designate the reagent as E (for electrophile) or N (for nucleophile), the reaction would proceed as outlined below.
| Reagent | Electrophile | Nucleophile | Typical Conditions |
| HBr | H+ | Br– | Low Temp |
| H20 | H+ | H2O (with loss of H+ after addition) | Aqueous acid/Low Temp |
| ROH | H+ | ROH | ROH/H3O+ |
| Br2 | Br+ | Br– | Br2/CCl4 |
| BrOH | Br– | –OH | Br2/H2O |
| BrOR | Br+ | –OR | Br2/ROH |
The intermediate carbocation is the tertiary carbocation, (rather than the primary carbocation that would be produced by addition to the =CH2 end of the double bond). This pattern of reaction is referred to as Markovinkov addition, after the person[1] who first discovered that HBr adds in this way to a double bond. We can classify many reagents as combinations of electrophile and nucleophile and, in this way, predict how they will add across the double bond. Examples of such reagents are shown (↑). Rather than memorizing the product of every type of addition across a double bond, it is much more productive to write a mechanism by determining which part is the electrophile, adding it to give the most stable carbocation, followed by the nucleophile.
Additions to alkenes are reversible: Let us now take a closer look at the addition of water across a double bond. Such a reaction can be accomplished by reacting the alkene with dilute sulfuric acid at low temperatures. The first step is addition of a proton to produce the most stable carbocation—which is then attacked by water (the nucleophile). The final product is the alcohol that forms after a proton is transferred to water. In this case, we can consider the proton (or more accurately H3O+) as a catalyst since it is regenerated at the end of the reaction sequence.
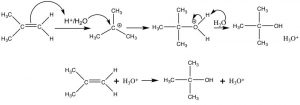
Acid-catalyzed addition of water across a double bond
At this point you might be asking yourself: well didn’t we just talk about the reverse reaction—that is, the elimination of H2O from alcohols to give alkenes? Indeed we did! Many organic reactions are reversible[2], it is just a matter of manipulating the conditions. The exact reaction conditions will determine which reaction is favored.
As is the case with most addition reactions, the addition of water across an alkene is exothermic, that is, ΔH is negative because stronger (sigma) bonds are formed during the reaction and energy is released into the environment. A typical energy diagram is shown below.
Reaction energy diagram for addition/elimination across a double bond.
This means that ΔH for the elimination reaction must be positive (i.e. going from right to left on the diagram above). The question then is: why does an elimination reaction ever occur? To answer that, we have to recall that the thermodynamic criterion for a reaction to proceed is not simply a negative enthalpy change, but rather a negative change in the Gibbs change (ΔG). Recall that ΔG = ΔH – TΔS. 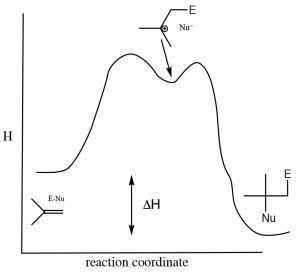 The change in entropy also influences the thermodynamic favorability of a reaction. In the elimination reaction, two molecules (alkene and water) are produced from one alcohol molecule; the entropy change will be positive. (Recall that entropy is associated with the number of possible arrangements of the system. Since two molecules will have more possible arrangements than one, this reaction will always be accompanied by an increase in entropy of the system.) To encourage the equilibrium to shift to the right (the addition reaction) we need increase the temperature, which will increase the magnitude of the –TΔS term, making ΔG more negative (assuming that ΔS is positive). In general, additions to double bonds are carried out at lower temperatures, while elimination reactions involve heating the reaction solution. Another way to influence the equilibrium state is to change the relative concentrations of reactants or products. Because water is a reactant, increasing the concentration of water shifts the equilibrium position towards the addition product while lowering the water concentration favors the elimination reaction.
The change in entropy also influences the thermodynamic favorability of a reaction. In the elimination reaction, two molecules (alkene and water) are produced from one alcohol molecule; the entropy change will be positive. (Recall that entropy is associated with the number of possible arrangements of the system. Since two molecules will have more possible arrangements than one, this reaction will always be accompanied by an increase in entropy of the system.) To encourage the equilibrium to shift to the right (the addition reaction) we need increase the temperature, which will increase the magnitude of the –TΔS term, making ΔG more negative (assuming that ΔS is positive). In general, additions to double bonds are carried out at lower temperatures, while elimination reactions involve heating the reaction solution. Another way to influence the equilibrium state is to change the relative concentrations of reactants or products. Because water is a reactant, increasing the concentration of water shifts the equilibrium position towards the addition product while lowering the water concentration favors the elimination reaction.

Specific reagents for additions across a double bond that reduce the carbocation problem
The problem with many of these simple addition reactions to a double bond is that they generate carbocations, which as we have seen already can lead to further reactions, resulting in skeletal rearrangements and the production of racemic mixtures (rather than a single stereoisomer). To address this issue, a number of reagents have been developed that minimize this problem. For example, a reagent that involves mercuric acetate (Hg(OAc)2) and sodium borohydride (NaBH4) as an intermediate can be used to add H2O, (or alcohol) across a double bond (↓).
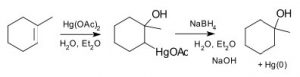
The reaction involves a mercury-stabilized cation (→) that prevents unwanted rearrangements. 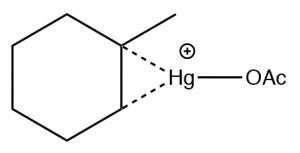 The product is still a Markovnikov product (see above) but is often formed more cleanly, that is, without unwanted alternatives.
The product is still a Markovnikov product (see above) but is often formed more cleanly, that is, without unwanted alternatives.
Another set of reactions that can be used to constrain molecular rearrangements and lead to stereospecific products are those that begin with the addition of bromine across the double bond. The simplest of these co-reactions is addition of Br2 itself; since Br is a large polarizable atom, the bromine molecule can become polarized and interact with the double bond as shown (↓) to form a bromonium ion (rather than a carbocation).
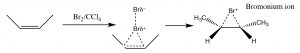
The bromonium ion can now undergo nucleophilic attack at either carbon (since in this example they are the same, that is, they are attached to identical groups), to produce the trans-dibromo addition product. The trans product is formed because the second step is an SN2 reaction with the bromide nucleophile attacking the carbon from the back-side.
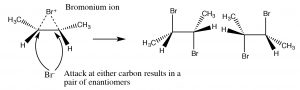
Addition of Br2 is accomplished by using a reaction solvent such as carbon tetrachloride that does not interfere with the reaction. If water or an alcohol is used as the solvent, then attack on the bromonium ion comes from the solvent acting as the nucleophile in the second step.
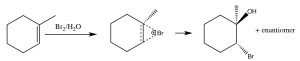
Again, the addition is trans, but now an incoming nucleophile (H2O) will attack the carbon that is most carbocation-like, that is it is the most stabilized, as shown here [latex]\rightarrow[/latex]. The reaction is both regiospecific and stereospecific. 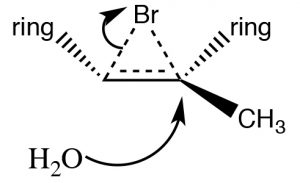
“Anti-Markovnikov” addition across double bonds
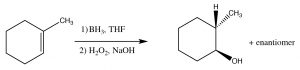
While the heading for this section is called “anti-Markovnikov” addition, this does not mean that the reaction mechanism is actually different. In the two examples we will discuss here, the difference is merely that the first addition to the double bond is not the H, which as we will see makes it appear that we have added a particular reagent the opposite way to the normal addition. 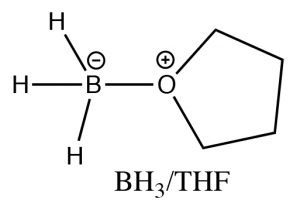 For example, if we want to add water across the double bond in to give the anti-Markovnikov product a different set of reagents is used: a Lewis acid-base complex of BH3 and the ether tetrahydrofuran (THF), followed by a solution of hydrogen peroxide in base. This reagent adds across the double bond in the direction that you would expect, that is the electrophile (Lewis acid) boron adds to the least substituted carbon, but at the same time, a hydrogen adds to the most substituted carbon from the same side of the molecule.
For example, if we want to add water across the double bond in to give the anti-Markovnikov product a different set of reagents is used: a Lewis acid-base complex of BH3 and the ether tetrahydrofuran (THF), followed by a solution of hydrogen peroxide in base. This reagent adds across the double bond in the direction that you would expect, that is the electrophile (Lewis acid) boron adds to the least substituted carbon, but at the same time, a hydrogen adds to the most substituted carbon from the same side of the molecule.
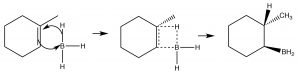
Mechanism of syn addition of BH3 across the double bond
This process happens twice more, and then the boron species is replaced by reaction with hydrogen peroxide and sodium hydroxide.
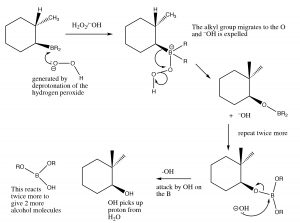
Mechanism of removal of boron moiety from the double bond
The overall reaction appears to have added the elements of water in an anti-Markovinkov direction. This reaction is not only regiospecific, but it is also stereospecific. The H and OH are added on the same (cis) side of the double bond and it is termed a syn addition.
Anti-Markovnikov addition of HBr across a double bond.
Another reaction which appears to violate what we have learned about the regiochemistry of addition across double bonds is the reaction of an alkene with HBr in the presence of light or peroxides. 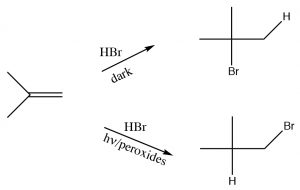 In contrast to the reaction we discussed previously, under conditions of light and in the presence of peroxides, the HBr adds in the reverse direction. Clearly something different is happening here: the reaction is proceeding by another Br mechanism. The clue is the presence of peroxides, which almost always signify that a reaction is proceeding via a radical mechanism rather than a polar mechanism.
In contrast to the reaction we discussed previously, under conditions of light and in the presence of peroxides, the HBr adds in the reverse direction. Clearly something different is happening here: the reaction is proceeding by another Br mechanism. The clue is the presence of peroxides, which almost always signify that a reaction is proceeding via a radical mechanism rather than a polar mechanism.
Radicals are species with unpaired electrons, and, as such, are very reactive. The reaction begins with an initiation step in which the peroxide (which contains a weak O–O bond) is broken homolytically to give two oxygen radicals. These react with HBr by abstracting a hydrogen, and leaving a bromine radical. Note that the oxy radical abstracts H and not Br, because Br is a more stable radical than H. Bromine radical is a large polarizable species and which can help stabilize the unpaired electron. A hydrogen radical is actually a hydrogen atom, it is highly unstable and reactive.
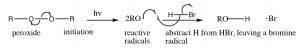
Note: when a mechanism involves single electrons moving (as in a homolytic bond cleavage, or any reaction of a radical species) we use what is called a fishhook arrow—with only one head, rather than the typical arrow that denotes movement of two electrons.
we use what is called a fishhook arrow—with only one head, rather than the typical arrow that denotes movement of two electrons.
The resulting bromine radical now reacts with the alkene double bond to produce the most stable intermediate, which is (just as in the carbocations) the tertiary. Carbon radicals show the same trends in stability as carbocations for the reason that they are also electron deficient and can be stabilized by the same mechanisms as carbocations (induction and hyperconjugation). The resulting carbon radical now abstracts an H from another molecule of HBr, to produce the anti-Markovnikov addition product, plus another bromine radical that can begin the cycle again. This is called a radical chain reaction—because it produces another reactive species that can continue the chain reaction.
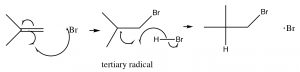
Note: Even though this reaction produces a different addition product than the typical addition of HBr across the double bond, the principles guiding the reaction are the same. The first addition produces the most stable intermediate; the difference is that bromine adds first.
Reduction of Alkenes:
The historical meaning of “reduction” involved reactions with hydrogen (H2), and conversely, oxidation meant reaction with oxygen (O2). This makes sense from the perspective that carbon is slightly more electronegative than hydrogen, so that a C-H bond is polarized as C∂– and H ∂+. Therefore, adding hydrogen to a C=C will increase (slightly) the negative charge on the carbon. (Similarly, a C-O bond is polarized C∂+ and O∂–, so that adding more oxygens to a carbon increases the amount of positive charge on the carbon.) Even today we refer to adding hydrogen across pi bonds as a reduction. However, alkenes do not normally react with hydrogen; typically a catalyst (usually a transition metal) is necessary for the reaction to occur. In general, the catalyst is supplied as a finely divided powder adsorbed onto an inert substance such as charcoal. The, most common catalysts are platinum or palladium on charcoal (Pt/C or Pd/C). 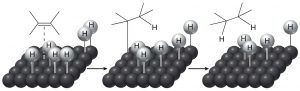 Typically, the substance to be reduced is dissolved in a solvent, the catalyst is added, and then hydrogen is bubbled through the mixture. The catalyst adsorbs both H2 and the alkene onto its surface and this interaction weakens both the H2 bond and the pi bond. The hydrogen then migrates to the adsorbed alkene and adds across the double bond. The reaction is stereospecific in that both H’s add from the same side—a syn addition. This can be seen more clearly if we use deuterium instead of hydrogen—both the D’s add from the same side.
Typically, the substance to be reduced is dissolved in a solvent, the catalyst is added, and then hydrogen is bubbled through the mixture. The catalyst adsorbs both H2 and the alkene onto its surface and this interaction weakens both the H2 bond and the pi bond. The hydrogen then migrates to the adsorbed alkene and adds across the double bond. The reaction is stereospecific in that both H’s add from the same side—a syn addition. This can be seen more clearly if we use deuterium instead of hydrogen—both the D’s add from the same side.

Oxidation of Alkenes
There are a variety of reagents that can result in the oxidation (i.e. the addition of oxygen to both carbons) of an alkene.[3] These reactions are synthetically useful because they enable us to place functional groups on adjacent carbons and these groups can subsequently be modified. The reagents used in these transformation reactions are highly reactive, and most include species in a high oxidation states, such as permanganate (MnO4–) and or Osmium tetroxide (OsO4), or contain unstable oxygen-oxygen bonds (e.g. Ozone O3) or a peroxy-acid (see below). The common factor in these reagents is that they are able to add oxygen in various ways to the C=C bond. Many of resulting reactions are quite complex, and we will not delve into their mechanistic details except where necessary: for example, to explain why a particular stereochemistry is produced.
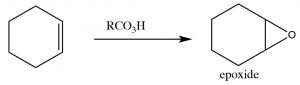 Epoxidation: Epoxides (also known as oxiranes) (→) are three-membered ring ethers, and can be formed by the reaction of an alkene with a per-acid, that is, a carboxylic acid with an extra oxygen (←).
Epoxidation: Epoxides (also known as oxiranes) (→) are three-membered ring ethers, and can be formed by the reaction of an alkene with a per-acid, that is, a carboxylic acid with an extra oxygen (←). 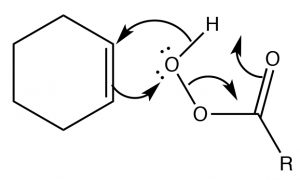 The reaction occurs via a concerted (coordinated) movement of electrons. The result is that both of the carbons in the original double bond end up linked to the same O atom.
The reaction occurs via a concerted (coordinated) movement of electrons. The result is that both of the carbons in the original double bond end up linked to the same O atom.
Recall that earlier we looked at relative stabilities of rings, and found that their stability depends on the ring size and the torsional (eclipsing) strain. A three membered carbon ring is highly strained because the bond angles are distorted away from the 109º angle that sp3 hybridization calls for; moreover, all of the bonds are eclipsed. 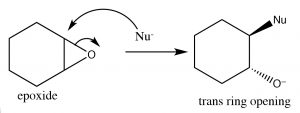 The result is that epoxides are susceptible to nucleophilic attack at a ring carbon (→). An SN2 reaction that proceeds via attack from the back side of the ring, leading to the production of the trans product. Such ring opening reactions can be accomplished by a range of nucleophiles, including water. The reaction with water results in a trans diol. In general, under SN2 conditions the ring opening is also stereospecific—that is the nucleophile will attack the least hindered carbon (↓).
The result is that epoxides are susceptible to nucleophilic attack at a ring carbon (→). An SN2 reaction that proceeds via attack from the back side of the ring, leading to the production of the trans product. Such ring opening reactions can be accomplished by a range of nucleophiles, including water. The reaction with water results in a trans diol. In general, under SN2 conditions the ring opening is also stereospecific—that is the nucleophile will attack the least hindered carbon (↓).
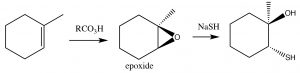
Epoxides tend to be reactive and for this reason can be useful as synthetic intermediates. Within biological systems, their reactivity can lead to chemical modification of DNA, leading to mutations (for that reason, many are known as genoxic or toxic to the genome). As a defense against such epoxides, organisms encode enzymes known as epoxide hydrolyzes.[4]
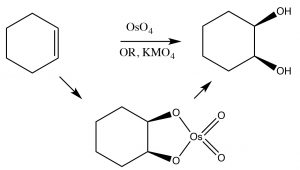 Cis-diols: Alkenes can be oxidized to produce cis-diols using a different type of reagent that adds atoms across the double bond via a cyclic intermediate. For example permanganate (MnO4–) and osmium tetroxide (OsO4), both of which contain transition metals in high-oxidation states, can accomplish this transformation (→). It is worth noting that by controlling the reaction conditions, we can choose to produce either cis or trans diols. As we move into more complex organic chemistry we will see that the ability to choose and predict outcomes is a major component of organic chemistry.
Cis-diols: Alkenes can be oxidized to produce cis-diols using a different type of reagent that adds atoms across the double bond via a cyclic intermediate. For example permanganate (MnO4–) and osmium tetroxide (OsO4), both of which contain transition metals in high-oxidation states, can accomplish this transformation (→). It is worth noting that by controlling the reaction conditions, we can choose to produce either cis or trans diols. As we move into more complex organic chemistry we will see that the ability to choose and predict outcomes is a major component of organic chemistry.
Ozonlysis: Another type of alkene double-bond oxidation involves a reaction with ozone (O3), the highly reactive allotrope of oxygen.[5] The mechanism is quite complex as shown below (no need to memorize it!).
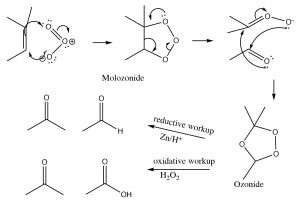
Typically, ozone cleaves the double bond and the reaction is treated with a mild reducing agent such as tin (Sn)[6], leading to the production of the corresponding aldehydes or ketones (↓).
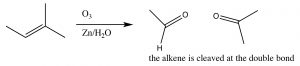
As we will see later, the ozonolysis reaction can be useful in identifying the position of a double bond within a molecule, as well as in the synthesis of aldehydes and ketones.
Reactions of Alkynes
As you might predict, alkynes often behave in a similar way to alkenes. The triple-bonded carbons are an electron-rich region of the molecule and we would expect them to undergo electrophilic addition, in a similar manner to alkenes. 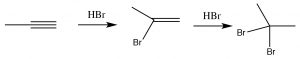 So, for example, we see Markovikov addition across the triple bond with HBr (→), the only difference being that if excess HBr is present, two—rather than one—bromine atom will be added; one to each of the originally triple-bonded carbons. Other reagents behave in a similar manner. For example Br2 will also add across the triple bond to give first the dibromo, and then the tetrabromo compound.
So, for example, we see Markovikov addition across the triple bond with HBr (→), the only difference being that if excess HBr is present, two—rather than one—bromine atom will be added; one to each of the originally triple-bonded carbons. Other reagents behave in a similar manner. For example Br2 will also add across the triple bond to give first the dibromo, and then the tetrabromo compound.
In contrast, when water is added across the triple bond we find a somewhat different outcome. 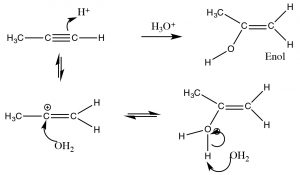 While the initial steps are the same: the electrophile (H+) adds to the least-substituted carbon, and the nucleophile (H2O) adds to the carbocation that is produced. This produces a new functionality called an enol (A combination of alkene and alcohol). The enol now undergoes what is known as a tautomerism: the proton from the alcohol moiety is removed (by water as a base), and another proton is picked up on the alkene CH2 carbon (→). As we have seen many times before this type protonation/deprotonation reaction occurs readily on either oxygen or nitrogen, but this is the first time we have seen it on a carbon; keto-enol tautomerism is an important part of the reactions of carbonyl groups.
While the initial steps are the same: the electrophile (H+) adds to the least-substituted carbon, and the nucleophile (H2O) adds to the carbocation that is produced. This produces a new functionality called an enol (A combination of alkene and alcohol). The enol now undergoes what is known as a tautomerism: the proton from the alcohol moiety is removed (by water as a base), and another proton is picked up on the alkene CH2 carbon (→). As we have seen many times before this type protonation/deprotonation reaction occurs readily on either oxygen or nitrogen, but this is the first time we have seen it on a carbon; keto-enol tautomerism is an important part of the reactions of carbonyl groups.
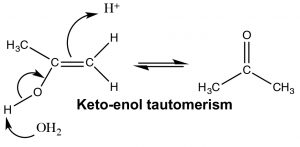
The keto and enol forms appear to be different compounds and we might be tempted to classify them as structural isomers—but they are not. The keto- and enol- forms always exist in an equilibrium with one another, and even though we usually write the structure with the carbonyl group (the keto form), there is always a small amount of the enol form present. The transition between keto- and enol- forms of the nucleotide bases initially confused Watson and Crick in their modeling of DNA structure.[7]
Reduction of alkynes: Addition of hydrogen (H2) to alkynes can be accomplished in several ways. It is possible to completely reduce the alkyne to the corresponding fully-saturated alkane through the addition of two H2 molecules. In fact, when using catalysts such as Pd (palladium) or Pt (platinum) the reaction cannot be stopped at the intermediate alkene stage. There are, however, specialized catalysts that allow for partial hydrogenation to the alkene. One of these is known as Lindlar’s catalyst, which is less efficient (poisoned) catalyst. As one might expect (by analogy with alkene reduction), the cis hydrogenated product is formed (↓).
![]() It is also possible to reduce an alkyne to the trans product, but to do this we have to use a different method; a method that involves the stepwise addition of the components of the reduction. The conditions for this reduction require a source of protons and a separate source of electrons. For example, a solution of sodium metal (a source of electrons) in liquid ammonia (a source of protons) at low temperature (since ammonia boils at -33ºC), can be used to reduce an alkyne, but since the reaction proceeds via a stepwise (not concerted) addition, the product formed is the trans alkene: the most stable product (→).
It is also possible to reduce an alkyne to the trans product, but to do this we have to use a different method; a method that involves the stepwise addition of the components of the reduction. The conditions for this reduction require a source of protons and a separate source of electrons. For example, a solution of sodium metal (a source of electrons) in liquid ammonia (a source of protons) at low temperature (since ammonia boils at -33ºC), can be used to reduce an alkyne, but since the reaction proceeds via a stepwise (not concerted) addition, the product formed is the trans alkene: the most stable product (→). ![]()
Acidity of Terminal Alkynes: One alkyne-specific reaction involves the acidity of protons attached to sp hybridized carbons. The pKa of such protons is around 25, which is much lower than that of alkanes (> 55) or alkenes (~ 45). In fact, “terminal” alkyne protons can be removed by strong bases such as NH2– (the amide ion), since the pKa of NH3 (ammonia) is 33 (↓).
![]() The acidity of terminal alkyne protons can be explained by the idea that the negative charge (the lone pair on the resulting anion) is located in an sp hybrid orbital. The more “s” character in the hybrid orbital, the closer to the nucleus. Since the sp orbital is more “s” than either sp2 or sp3 orbitals, then the electron of the carbon anion is held closer to the nucleus and is therefore more stable than if the carbon were sp2 or sp3 hybridized. The effect is similar to the effective nuclear charge explanation for the trends in electronegativity across the periodic table (i.e. why fluorine is more electronegative than oxygen). The alkyne anion is very useful because it is now a carbon nucleophile, and will attack electrophilic carbon species in an SN2 reaction. This is the first example we have seen of carbon carbon bond formation (although we will see many more).
The acidity of terminal alkyne protons can be explained by the idea that the negative charge (the lone pair on the resulting anion) is located in an sp hybrid orbital. The more “s” character in the hybrid orbital, the closer to the nucleus. Since the sp orbital is more “s” than either sp2 or sp3 orbitals, then the electron of the carbon anion is held closer to the nucleus and is therefore more stable than if the carbon were sp2 or sp3 hybridized. The effect is similar to the effective nuclear charge explanation for the trends in electronegativity across the periodic table (i.e. why fluorine is more electronegative than oxygen). The alkyne anion is very useful because it is now a carbon nucleophile, and will attack electrophilic carbon species in an SN2 reaction. This is the first example we have seen of carbon carbon bond formation (although we will see many more). ![]()
- https://en.wikipedia.org/wiki/Vladimir_Markovnikov ↵
- In fact ALL reactions are reversible in theory (this is called the principle of microscopic reversibility, https://en.wikipedia.org/wiki/Microscopic_reversibility . However, in practice it is extremely difficult to reverse some reactions in the laboratory. For example, combustion of hydrocarbons is not something you would try to reverse in the lab, since the products are gases and will be very difficult to bring back together, and the reaction is highly exergonic. However, plants can do the reverse reaction quite well using energy from sunlight. ↵
- While we have seen that alkenes can add water (as H+ and –OH) across a double bond, this is not classified as an oxidation. There is no change in oxidation state of the O or H that add to the double bonded carbons. ↵
- epoxide hydrolases: http://www.annualreviews.org/doi/pdf/10.1146/annurev.pharmtox.45.120403.095920 ↵
- Ozone is generated during the reaction by using a special generator because it is too reactive to store. It is generated in the same way that lightning generates ozone—by passing a spark of electric current through oxygen. ↵
- The reducing agent is present to stop “over oxidation” to the carboxylic acid. ↵
- Tautomers: evil twins of the bases!: http://blc.arizona.edu/courses/181Lab/MoBiByMe/Tautomers.html ↵
