
Chapter 8: Conjugated compounds and aromaticity
Conjugation: Conjugation is the term we use to describe an arrangement of alternating single and double bonds.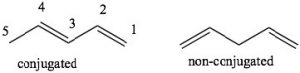 To explain how conjugated systems behave differently from non-conjugated systems we will compare 1,3-pentadiene (which is conjugated) and 1,4 pentadiene (which is not conjugated). To recognize the differences between the two, let us look at the orbitals that are involved in the π system bonding. Recall that π bonds can be considered as the side-to-side overlap of p orbitals, so that the electron density lies above and below the plane of the rest of the molecule. Consider the orbitals in 1,3-pentadiene: there is overlap of p orbitals that result in a continuous
To explain how conjugated systems behave differently from non-conjugated systems we will compare 1,3-pentadiene (which is conjugated) and 1,4 pentadiene (which is not conjugated). To recognize the differences between the two, let us look at the orbitals that are involved in the π system bonding. Recall that π bonds can be considered as the side-to-side overlap of p orbitals, so that the electron density lies above and below the plane of the rest of the molecule. Consider the orbitals in 1,3-pentadiene: there is overlap of p orbitals that result in a continuous
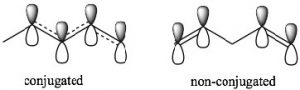 π orbital system over carbons 1-4. The consequence of this is that there is some partial double-bond character between carbons 2 and 3. In 1,4-pentadiene there is no possibility of overlap between the two separate π bonds. Note that in the non-conjugated system, there is an sp3 hybridized carbon between the two sets of sp2 hybridized carbon-carbon double bonds which prevents any overlap of (p) orbitals on carbons 2 and 4.
π orbital system over carbons 1-4. The consequence of this is that there is some partial double-bond character between carbons 2 and 3. In 1,4-pentadiene there is no possibility of overlap between the two separate π bonds. Note that in the non-conjugated system, there is an sp3 hybridized carbon between the two sets of sp2 hybridized carbon-carbon double bonds which prevents any overlap of (p) orbitals on carbons 2 and 4.
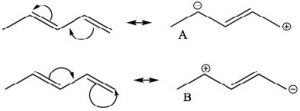 One way to indicate and predict where this partial double bond character can occur is to use resonance structures. We can write resonance structures for the conjugated system which have double bond character between C-2 and C-3. Note that since resonance contributors A and B are equivalent, there is no actual charge separation in this molecule. It is not possible (without breaking a sigma bond) to write resonance structures like this for 1,4-pentadiene (try and convince yourself that this is true ).
One way to indicate and predict where this partial double bond character can occur is to use resonance structures. We can write resonance structures for the conjugated system which have double bond character between C-2 and C-3. Note that since resonance contributors A and B are equivalent, there is no actual charge separation in this molecule. It is not possible (without breaking a sigma bond) to write resonance structures like this for 1,4-pentadiene (try and convince yourself that this is true ).
Molecular Orbital Theory: Another model that can be used to describe the bonding is to consider the π system in terms of molecular orbital theory. Molecular orbital theory considers the bonding orbitals as extending over the whole molecule. The number of molecular orbitals is equal to the sum of all the atomic orbitals. In practice, this approach is far too complex for even the smallest of organic molecules, which is why we usually use the simpler valence bond model in which we consider bonds as being located between two atoms. In the case of conjugated systems, it is often helpful to use the valence bond approach for the sigma (single bonds) framework and then consider the conjugated π system using molecular orbital theory. In this case, if we have a conjugated system of two π bonds, then four p atomic orbitals are involved in forming the four molecular orbitals (MOs). Recall that when molecular orbitals are formed from atomic orbitals (AOs), if the (quantum mechanical) wave functions add in phase, the resulting energy of the MO is lower—that is, the interaction is stabilizing and the MOs are bonding MOs. If the AOs add out of phase, the interaction is destabilizing and the result is antibonding MOs. Note that, in the diagram (↓), only the lowest energy MO has electron density between carbons 2 and 3. All the other MOs have a node (no electron density) at this position. Overall, we see that there is more π electron density between C-1 and C-2, and between C-3 and C-4.
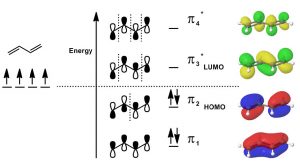
Since there are only 4 π electrons, only the two bonding MOs are occupied while the two antibonding MOs are unoccupied. As we will see, if we consider reactions using MO theory, the Highest Occupied MO (HOMO) and the Lowest Unoccupied MO (LUMO) are the orbitals that participate in new bonding interactions. In general, we use the simplest model that allows us to predict and explain the outcome of reactions, which is usually valence bond theory, but we will call on MO theory when necessary—and discussions of conjugated systems sometimes require MO theory to explain phenomena.
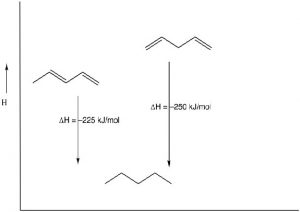
Stability of Conjugated Systems: As we have seen before, one way to identify the thermodynamic stability of alkenes is to reduce them to the corresponding alkane by adding H2 (hydrogens) across the double bond and determine the enthalpy change.[1] In the case of 1,3- and 1,4-pentadiene, we can compare their heats of hydrogenation to produce the corresponding pentane; we find that the conjugated diene is about 25 kJ/mol more stable. This is a general finding: the more conjugated a system, the more stable it is and the less reactive it is.
UV-VIS Spectroscopy and Conjugated Systems: Review[2]
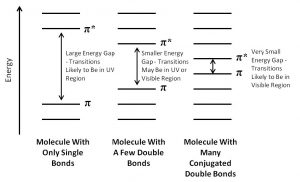
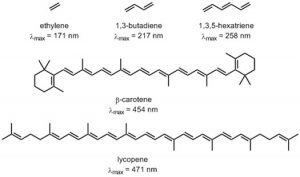
As you probably remember, the more atomic orbitals that combine to produce molecular orbitals, the smaller the energy gap between the MOs becomes. For example, an electron in an isolated pi bond absorbs energy in the far UV (~170 nm), a rather high energy. As we increase the number of conjugated double bonds, the energy gap between the orbitals (in fact the gap between the HOMO and LUMO) gets smaller and smaller, so that lower energy photons can bring about this transition. Eventually, the wavelength of light needed to promote an electron from highest occupied to the lowest unoccupied MO (HOMO→LUMO) moves into the visible region, and the substance becomes colored. Note that its color does not represent the the light that is absorbed, but rather the light that is transmitted or reflected. These conjugated regions of molecules are called chromophores.[3] The longer the conjugated section of the molecule, the longer the wavelength that is absorbed. The compounds responsible for highly colored fruits and vegetables—such as lycopene and B-carotene, as well as your ability to see visible light (retinals) contain large chromophore regions.
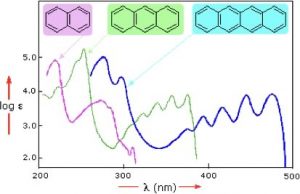 Samples of UV-VIS absorption spectra are shown here (→). Note that, in contrast with most other spectroscopic techniques (which usually produce sharp lines), the peaks in these spectra are more broad; the longer the conjugated section of the chromophore is, the longer the wavelength (and lower energy) that it absorbs. This means that each of these compounds has a different color. Moreover, the fact that the peaks in these spectra are not sharp means that UV-VIS spectroscopy is typically not used for identification of compounds. However, the amount of light absorbed is proportional to the concentration of the substance, so UV-VIS spectroscopy can be used to determine the concentration of samples. The visible spectrum runs from about 300nm to 750nm.
Samples of UV-VIS absorption spectra are shown here (→). Note that, in contrast with most other spectroscopic techniques (which usually produce sharp lines), the peaks in these spectra are more broad; the longer the conjugated section of the chromophore is, the longer the wavelength (and lower energy) that it absorbs. This means that each of these compounds has a different color. Moreover, the fact that the peaks in these spectra are not sharp means that UV-VIS spectroscopy is typically not used for identification of compounds. However, the amount of light absorbed is proportional to the concentration of the substance, so UV-VIS spectroscopy can be used to determine the concentration of samples. The visible spectrum runs from about 300nm to 750nm.
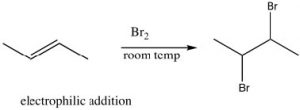
Reactions of Conjugated Systems: Kinetic and Thermodynamic Control of Reactions: Now let us consider the reaction of 1,3-butadiene with a reagent such as HCl. Just as we saw with isolated alkenes, the first step in the reaction is the addition of the electrophile H+ to produce the most stable carbocation. In this reaction, the proton adds to C-1; the resulting carbocation is resonance-stabilized with positive charge at both C-2 and C-4. The question, then, is: where does the nucleophile add?
![]() In fact, the position of equilibrium depends upon the conditions under which the reaction occurs. First, let us consider the two potential sites of attack. Attack at C-2 would mean that the reagent has added across the C-1/C-2 pi bond—this is called 1,2 addition.
In fact, the position of equilibrium depends upon the conditions under which the reaction occurs. First, let us consider the two potential sites of attack. Attack at C-2 would mean that the reagent has added across the C-1/C-2 pi bond—this is called 1,2 addition. ![]() Attack at C-4 is called 1,4-addition and the two products are clearly different.
Attack at C-4 is called 1,4-addition and the two products are clearly different.
The partial positive charge at C-2 is located on a secondary carbon, whereas that on the C-4 is on a primary carbon. This means that the intermediate carbocation has more partial positive charge on C-2 than on C-4 and the transition state for the reaction to give the 1,2-addition product will have a lower activation energy than the transition state for the 1,4 product because it is more stabilized (by induction and hyper-conjugation). Therefore, the attack of the nucleophile (chloride) would occur faster at C-2; that is, 1,2 addition is the kinetically favored product.
However, if we consider the 1,4-addition product, the alkene product itself is more substituted (there are two alkyl groups on the double bond) and, therefore, it is more stable than the product of 1,2 addition. Even though the 1,4-addition product is formed more slowly through a less-stabilized transition state, the product itself is more stable; it is the thermodynamically favored product.
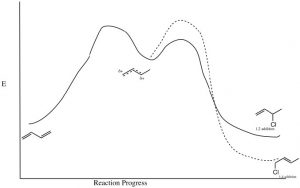
In fact, by controlling the reaction conditions, it is possible to produce either the kinetic or the thermodynamic product. If the reaction is run at relatively low temperature, there will not be enough energy to overcome the activation energy barrier associated with the 1,4-addition product reaction and the kinetic (1,2) product will be produced. However, at higher temperatures, there is enough energy; the 1,4 activation energy barrier will be reached more often, so that even if the kinetic product is formed, the fact that the reaction is reversible will, over time, lead to the most stable product accumulating at equilibrium. It is important to note that the most stable product is not always formed—it depends upon reaction conditions.
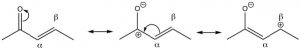
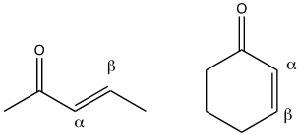 Conjugated carbonyl compounds: Carbonyl compounds can also be conjugated with carbon-carbon double bonds. These compounds are often referred to as α,β unsaturated carbonyls (the carbon next to the carbonyl carbon is termed the alpha carbon). By drawing resonance forms for this system, we see that there is a partial positive charge on the carbonyl carbon and on the β carbon. This means that α,β unsaturated carbonyl compounds are susceptible to nucleophilic attack at both the C=O and at the β carbon. Which is analogous to the 1,2- and 1,4-additions to conjugated dienes.
Conjugated carbonyl compounds: Carbonyl compounds can also be conjugated with carbon-carbon double bonds. These compounds are often referred to as α,β unsaturated carbonyls (the carbon next to the carbonyl carbon is termed the alpha carbon). By drawing resonance forms for this system, we see that there is a partial positive charge on the carbonyl carbon and on the β carbon. This means that α,β unsaturated carbonyl compounds are susceptible to nucleophilic attack at both the C=O and at the β carbon. Which is analogous to the 1,2- and 1,4-additions to conjugated dienes.
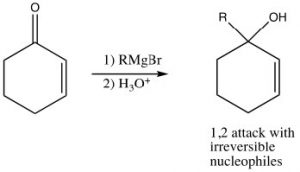 Just as the 1,3-conjugated diene case, attack at the carbonyl carbon is the kinetically preferred product since there is more positive charge there and the reaction is faster. Reagents that attack carbonyls irreversibly, such as grignards, alkyl lithiums, or reducing agents such as LiAlH4 will, therefore, tend to produce the product of attack at the carbonyl. This is the 1,2-addition product.
Just as the 1,3-conjugated diene case, attack at the carbonyl carbon is the kinetically preferred product since there is more positive charge there and the reaction is faster. Reagents that attack carbonyls irreversibly, such as grignards, alkyl lithiums, or reducing agents such as LiAlH4 will, therefore, tend to produce the product of attack at the carbonyl. This is the 1,2-addition product.
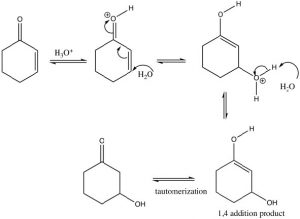 If we use a reversible nucleophile (ROH, H2O, RNH2), then the most thermodynamically stable product will predominate, that is, the product of 1,4 addition. However, this product undergoes a tautomerism[4] that regenerates the carbonyl (which is the source of the stability—recall C=O bonds are very strong).
If we use a reversible nucleophile (ROH, H2O, RNH2), then the most thermodynamically stable product will predominate, that is, the product of 1,4 addition. However, this product undergoes a tautomerism[4] that regenerates the carbonyl (which is the source of the stability—recall C=O bonds are very strong).
Another reagent that can produce a product resulting from attack at the beta carbon is a reagent that we have not seen yet, an organocuprate, otherwise known as the Gilman[5] reagent, which consists of a complex of alkyl groups, copper, and lithium and has the generic formula R2CuLi. The general reaction is shown here. The organocuprate has the two alkyl groups bonded to the copper species (formally Cu2+), 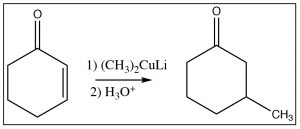 but the carbon-copper bond is more covalent than ionic (the charge on the carbon is lower than on the equivalent Grignard or alkyl lithium reagent) and this makes attack at the beta carbon more likely.[6]
but the carbon-copper bond is more covalent than ionic (the charge on the carbon is lower than on the equivalent Grignard or alkyl lithium reagent) and this makes attack at the beta carbon more likely.[6]
![]() Gilman reagents are also very useful because they can accomplish nucleophilic attack on alkyl halides, which does not occur with Grignard or alkyl lithium reagents.
Gilman reagents are also very useful because they can accomplish nucleophilic attack on alkyl halides, which does not occur with Grignard or alkyl lithium reagents.
Aromaticity
After considering some of the properties of conjugated systems, we now move on to what might be the ultimate in conjugated systems, as exemplified by benzene C6H6. While many of the properties of benzene and its derivatives are similar in some ways to those of open-chained conjugated systems, there are important differences. Benzene has the property known as aromaticity and we say that benzene is aromatic. In everyday language, the term aromatic implies that something smells; usually in a good way. While benzene does have a fairly strong smell[7], in chemistry, aromatic has come to mean a particular set properties that emerge from the molecular structure of some molecules. 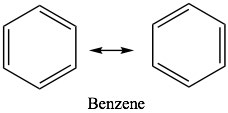 Benzene is the simplest and most common example of an aromatic compound. The structure of benzene was something of a puzzle for quite a long time; it eventually came to be written in the form of what are now called Kekulé structures in which double and single bonds appear to alternate around the ring. We can write two equivalent resonance structures which contribute equally to the overall structure of the molecule. While these models can serve us well when trying to figure out what the electrons are doing during reactions, neither is not adequate to represent the actual structure of benzene.
Benzene is the simplest and most common example of an aromatic compound. The structure of benzene was something of a puzzle for quite a long time; it eventually came to be written in the form of what are now called Kekulé structures in which double and single bonds appear to alternate around the ring. We can write two equivalent resonance structures which contribute equally to the overall structure of the molecule. While these models can serve us well when trying to figure out what the electrons are doing during reactions, neither is not adequate to represent the actual structure of benzene. 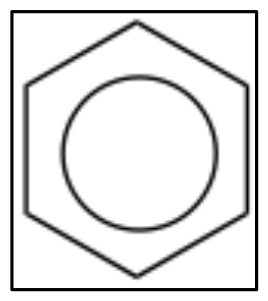 Sometimes, you will see benzene written with a circle in the middle (ß) to indicate that, in reality, there are no single or double bonds present; rather, there is the same electron density (1.5 bonds) between all the carbons.
Sometimes, you will see benzene written with a circle in the middle (ß) to indicate that, in reality, there are no single or double bonds present; rather, there is the same electron density (1.5 bonds) between all the carbons.
Benzene has some rather remarkable properties which led chemists to classify it as a member of a completely different type of functionality. For example, benzene is much more stable than one might imagine—even for a conjugated system. The heat of hydrogenation of benzene is -208 kJ/mole, while the ΔH of hydrogenation of the isolated double bond in cyclohexene is 130 kJ/mol. We can see the effects of conjugation in 1,3-cyclo-hexadiene, which is 232 kJ/mol (not 260 as might be expected if it were conjugated). Similarly, if benzene were not conjugated, we would expect a heat of hydrogenation to be 3×130 = 390 kJ/mol. Therefore, the difference between the expected and the actual hydrogenation energy must be due to the stabilization conferred by resonance.
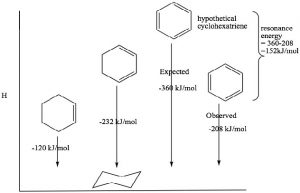
 This stability is called the resonance energy and, in aromatic compounds like benzene, it has a significant effect on the properties of the substance and types of reactions that a molecule participates in. For example, benzene does not react with electrophiles in the same way as isolated alkenes or even open-chained conjugated alkenes. Recall that the most common reactivity of simple alkenes is the electrophilic addition of E-Nu, where E is an electrophile and Nu is the nucleophile, across the double bond. In contrast, benzene undergoes electrophilic substitution; typically the reaction conditions require a catalyst and extensive heating.
This stability is called the resonance energy and, in aromatic compounds like benzene, it has a significant effect on the properties of the substance and types of reactions that a molecule participates in. For example, benzene does not react with electrophiles in the same way as isolated alkenes or even open-chained conjugated alkenes. Recall that the most common reactivity of simple alkenes is the electrophilic addition of E-Nu, where E is an electrophile and Nu is the nucleophile, across the double bond. In contrast, benzene undergoes electrophilic substitution; typically the reaction conditions require a catalyst and extensive heating. 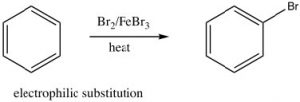 We will discuss the mechanism of this reaction shortly, but for now the important thing to note is that it is difficult to disrupt the aromatic ring of electrons and, when that does happen, the aromatic ring regenerates.
We will discuss the mechanism of this reaction shortly, but for now the important thing to note is that it is difficult to disrupt the aromatic ring of electrons and, when that does happen, the aromatic ring regenerates.
What is aromaticity and how do we recognize aromatic systems?
 To recap, benzene is uniquely stable and it is relatively difficult to get it to react, despite the high electron density in the ring. It is significantly more stable than cyclohexadiene which is conjugated, but only contains two bonds. There is clearly something special about a conjugated ring system above and beyond simple conjugation properties.
To recap, benzene is uniquely stable and it is relatively difficult to get it to react, despite the high electron density in the ring. It is significantly more stable than cyclohexadiene which is conjugated, but only contains two bonds. There is clearly something special about a conjugated ring system above and beyond simple conjugation properties.
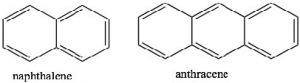
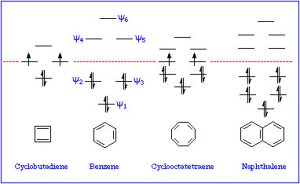
 If we examine other systems, we can begin to find the parameters that govern the property of aromaticity. We can begin by looking at some other cyclic, conjugated systems. Many fused six-membered rings (for example: naphthalene and anthracene) are aromatic. However, cyclobutadiene and cyclooctatetraene are not aromatic and have quite different properties.
If we examine other systems, we can begin to find the parameters that govern the property of aromaticity. We can begin by looking at some other cyclic, conjugated systems. Many fused six-membered rings (for example: naphthalene and anthracene) are aromatic. However, cyclobutadiene and cyclooctatetraene are not aromatic and have quite different properties.
By investigating many cyclic conjugated systems, it has been possible to identify the factors that lead to aromaticity.
These are known as Huckel’s Rule: Aromatic compounds are planar, cyclic, conjugated, and have 4n+2 π electrons in the π electron cloud. Benzene is the archetypal aromatic compound: it is planar (all carbons sp2 hybridized), cyclic (obviously) conjugated (apparent alternating single and double bonds), and it has 6 π electrons (n=1). If we look at the bonding within benzene, we see that overlapping p orbitals form a ring of pi electron density above and below the sigma C-C framework. 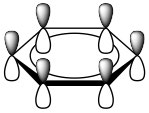 More generally, aromatic compounds must be planar, cyclic, and conjugated so that the p orbitals overlap to form this continuous ring of electron density. So why do aromatic compounds have 4n+2 π electrons? The answer involves molecular orbital theory.
More generally, aromatic compounds must be planar, cyclic, and conjugated so that the p orbitals overlap to form this continuous ring of electron density. So why do aromatic compounds have 4n+2 π electrons? The answer involves molecular orbital theory.
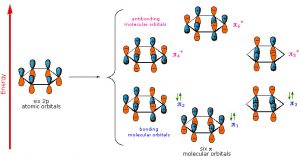
As previously noted, a consideration of the number and type of atomic orbitals that contribute to the bonding system enables MO theory to predict the molecular orbitals that span the whole molecule. In benzene, we have six p atomic orbitals and, therefore, expect that they combine to give six molecular orbitals as shown here. Note that there are three bonding and three antibonding MOs and, since there are only six electrons in the system, we get a total of three bonds.
This type of analysis can be done for any cyclic conjugated system. While it is too complex here to go into the mathematical underpinnings, there is a relatively simple way to determine the relative energies of the MOs. The approach requires that you inscribe the cyclic system into a circle, with one corner of the ring at the bottom. The places where the corners meet the ring are represent the relative energies of the MOs, as shown below. This arrangement is the origin of the 4n+2 rule as we will see.
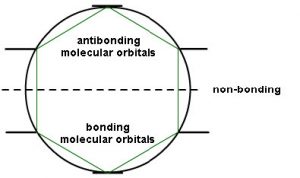
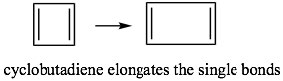 So, for example, if we consider cyclobutadiene (which has 4n π electrons), we see that there are 4 MOs and 4 electrons. However, two of those electrons are in non-stabilized orbitals AND are uncoupled because the orbitals are of the same energy. Remember Hund’s rule: electrons occupy orbitals singly until they have to start doubling up.
So, for example, if we consider cyclobutadiene (which has 4n π electrons), we see that there are 4 MOs and 4 electrons. However, two of those electrons are in non-stabilized orbitals AND are uncoupled because the orbitals are of the same energy. Remember Hund’s rule: electrons occupy orbitals singly until they have to start doubling up.
This means that if cyclobutadiene were aromatic, it would be highly unstable because it would contain two unpaired electrons—it would exist as a diradical. The presence of unpaired electrons generally makes compounds very reactive. In fact, cyclobutadiene is not aromatic and instead exists as two isolated double bonds by lengthening the single bonds. Even so, it is still unstable and does not exist above -78º C.
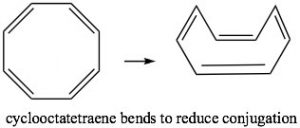 Cyclooctatetraene also has 4n π electrons (where n=2), and again we see the problem is that there are a pair of degenerate (same energy) MOs, where the last two π electrons are located. Again, this is highly destabilizing and, to avoid this electron configuration, cyclooctatetraene actually bends so the double bonds are not conjugated with each other. The origin of the n+2 rule, then, has to do with the arrangement of MOs. Any cyclic conjugated compound with 4n π electrons will not be able to take up a stable conjugated arrangement because it will involve the highly unstable diradical.
Cyclooctatetraene also has 4n π electrons (where n=2), and again we see the problem is that there are a pair of degenerate (same energy) MOs, where the last two π electrons are located. Again, this is highly destabilizing and, to avoid this electron configuration, cyclooctatetraene actually bends so the double bonds are not conjugated with each other. The origin of the n+2 rule, then, has to do with the arrangement of MOs. Any cyclic conjugated compound with 4n π electrons will not be able to take up a stable conjugated arrangement because it will involve the highly unstable diradical.
Aromatic Ions
The stability conveyed by aromaticity can be a powerful driving force. For example, cyclopentadiene is quite acidic with a pKa of 15 (compared to > 50 for alkanes). It is the sp3 hybridized carbon that is deprotonated and it rehybridizes to sp2 so that conjugation around the ring is possible. The product cyclopentadienyl anion is aromatic since it now has 6 π electrons; two from the lone pair resulting from the deprotonation and four from the original pi system. It is the drive towards aromaticity that makes cyclopentadiene so much more acidic than a normal alkene.
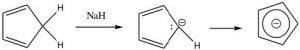
Similarly, cycloheptatriene can be induced to become a positively charged aromatic ion by treating the corresponding alcohol with acid which will allow the OH to leave as H2O, 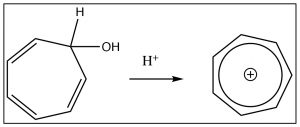 leaving behind a positively charged delocalized aromatic ion (the tropylium cation), which has six electrons delocalized over seven carbon atoms. In fact, there are quite a few ways to generate such aromatic anions, and they are all more stable than might be predicted if you didn’t know about aromaticity.
leaving behind a positively charged delocalized aromatic ion (the tropylium cation), which has six electrons delocalized over seven carbon atoms. In fact, there are quite a few ways to generate such aromatic anions, and they are all more stable than might be predicted if you didn’t know about aromaticity.
Heterocyclic aromatic compounds
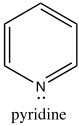 While the aromatic ions are interesting curiosities, there is a class of aromatic compounds that are of practical importance from a biological perspective: the heterocyclic aromatic compounds. These compounds typically have one or more carbons replaced by N, O, or S. For example, pyridine can be considered as the nitrogen analog of benzene.
While the aromatic ions are interesting curiosities, there is a class of aromatic compounds that are of practical importance from a biological perspective: the heterocyclic aromatic compounds. These compounds typically have one or more carbons replaced by N, O, or S. For example, pyridine can be considered as the nitrogen analog of benzene. 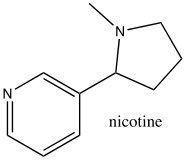 The nitrogen is sp2 hybridized and contributes an electron to the aromatic system. Pyridine is an important component of many biological molecules; for example, nicotine, a component of the NAD and NADH oxidation/reduction system discussed earlier. The lone pair on the pyridine nitrogen sits in an sp2 hybrid orbital at right angles to the pi system (like the C-H bonds) and, therefore, pyridine is basic because the lone pair is accessible. In fact, pyridine is often used as a base to react with any by-product acid such as HCl that might be produced in a reaction.
The nitrogen is sp2 hybridized and contributes an electron to the aromatic system. Pyridine is an important component of many biological molecules; for example, nicotine, a component of the NAD and NADH oxidation/reduction system discussed earlier. The lone pair on the pyridine nitrogen sits in an sp2 hybrid orbital at right angles to the pi system (like the C-H bonds) and, therefore, pyridine is basic because the lone pair is accessible. In fact, pyridine is often used as a base to react with any by-product acid such as HCl that might be produced in a reaction.
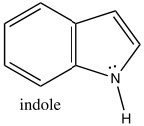 Indole is another heterocyclic aromatic system, but, in contrast to pyridine, indole uses the nitrogen lone pair to contribute the aromatic pi system. Indole has 10 π electrons (n=2), of which two are from the nitrogen lone pair.
Indole is another heterocyclic aromatic system, but, in contrast to pyridine, indole uses the nitrogen lone pair to contribute the aromatic pi system. Indole has 10 π electrons (n=2), of which two are from the nitrogen lone pair.
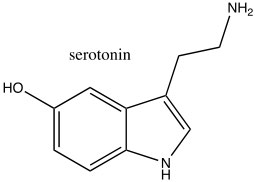
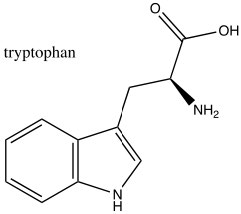
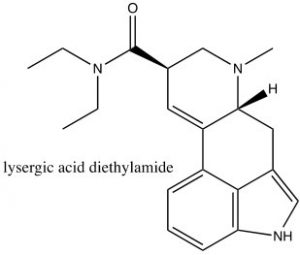
Consequently, indole is not basic. The ring system of indole also appears in many biologically important molecules, including the neurotransmitter serotonin, amino acid tryptophan, and hallucinogen lysergic acid diethylamide (LSD).
There are many biologically important nitrogenous aromatic 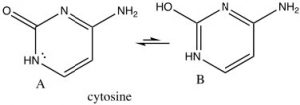 heterocycles, for example the bases in DNA and RNA all contain heterocyclic aromatic rings. For example, cytosine, which is usually written in the keto form (structure A) – which may not at first sight seem to be aromatic. However, all the atoms in the ring are planar sp2 hybridized and there are six electrons in the ring. The result is that cytosine can exist in its tautomeric enol form (B), but the keto form is actually more stable (because of the C=O). Both enol and keto forms are aromatic.
heterocycles, for example the bases in DNA and RNA all contain heterocyclic aromatic rings. For example, cytosine, which is usually written in the keto form (structure A) – which may not at first sight seem to be aromatic. However, all the atoms in the ring are planar sp2 hybridized and there are six electrons in the ring. The result is that cytosine can exist in its tautomeric enol form (B), but the keto form is actually more stable (because of the C=O). Both enol and keto forms are aromatic.
Spectroscopy of aromatic compounds:
As you might expect, both the C-13 and the 1H NMR spectrum of benzene show only one peak (H-NMR 7.3 ppm and C-NMR 128 ppm) meaning that there is only one type of carbon and one type of hydrogen present in the molecule. However, these single peaks appear at lower field strengths relative to those found in alkenes or even conjugated alkenes (which normally appear between 5 and 6 ppm in the H-NMR). This low field absorption is caused by a phenomenon that occurs when the cyclic electron cloud in the ring is placed into an external magnetic field. The ring of electron density begins to cycle, producing a ring current and an induced magnetic field; the resulting intrinsic field reinforces the external field.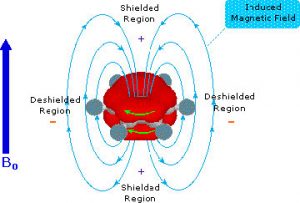 The result is that the external field does not need to be very high to bring the carbon or hydrogen nuclei to resonance. In effect, the aromatic carbons and hydrogens are deshielded and appear at low field.
The result is that the external field does not need to be very high to bring the carbon or hydrogen nuclei to resonance. In effect, the aromatic carbons and hydrogens are deshielded and appear at low field.
Interestingly, the induced field opposes the external field in the center of the ring, and, in fact, there are cyclic aromatic polyenes where (because of structural constraints) some of the hydrogens do point to the center of the ring. 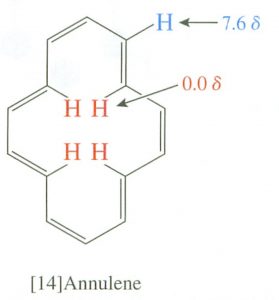 There is a marked difference between the inner and outer hydrogen resonances in these compounds because the hydrogens are in different areas of the induced magnetic field. This effect is called diamagnetic anisotropy.
There is a marked difference between the inner and outer hydrogen resonances in these compounds because the hydrogens are in different areas of the induced magnetic field. This effect is called diamagnetic anisotropy.
The IR spectra of aromatic compounds typically show a C-H stretch above 3,000 cm-1, and a C-C bend around 1600 cm-1, but these signals are often mixed in with others and, in general, IR is not very useful for identifying aromatic compounds. On the other hand, UV-VIS spectroscopy is often used to identify the aromatic chromophore. Benzene itself has a broad absorption around 254 nm and, as we will see, this absorption changes depending on the electron withdrawing and donating properties of groups on the ring itself.
Reactions of Aromatic Compounds: Introduction of one group onto the ring
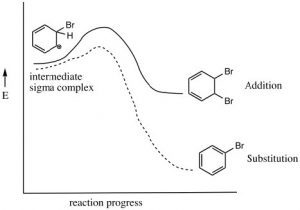
As we have seen, aromatic compounds are considerably more stable than one might predict. Consequently, it takes more energy to make aromatic compounds undergo reactions since, in order to react the aromatic overlap of orbitals in the ring, it must be destroyed at some point during the reaction. 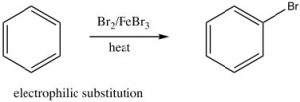 Since the aromatic ring is so electron-rich we might predict that it would undergo electrophilic attack but, rather than undergoing electrophilic addition like alkenes and conjugated alkenes do, aromatic compounds typically undergo electrophilic substitution. For example, benzene will react with bromine in the presence of a catalyst such as iron (III) bromide (FeBr3) to give bromobenzene.
Since the aromatic ring is so electron-rich we might predict that it would undergo electrophilic attack but, rather than undergoing electrophilic addition like alkenes and conjugated alkenes do, aromatic compounds typically undergo electrophilic substitution. For example, benzene will react with bromine in the presence of a catalyst such as iron (III) bromide (FeBr3) to give bromobenzene.
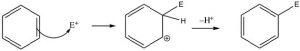
Let us now take a closer look at the steps involved in this reaction to see how this substitution (Br for H) is accomplished. ![]() Since benzene is so stable, a more reactive electrophile is needed to react with the ring. This is accomplished by adding a Lewis acid catalyst FeBr3, which forms a complex with the bromine to produce Br+ (stabilized by the FeBr4–). The Br+ electrophile now reacts with the electron-rich benzene ring to produce a resonance-stabilized intermediate called a sigma complex.
Since benzene is so stable, a more reactive electrophile is needed to react with the ring. This is accomplished by adding a Lewis acid catalyst FeBr3, which forms a complex with the bromine to produce Br+ (stabilized by the FeBr4–). The Br+ electrophile now reacts with the electron-rich benzene ring to produce a resonance-stabilized intermediate called a sigma complex.
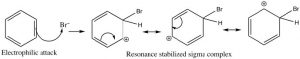
Now, instead of a nucleophile attacking, the aromatic ring is regenerated by loss of a proton.

The resulting bromobenzene is much more stable than the corresponding addition product.
This electrophilic substitution reaction is the primary mechanism by which most aromatic compounds react. A very reactive electrophile must be generated; it then adds to one of the ring carbons followed by loss of a proton from the same carbon.
There are a number of substituents that can be introduced onto the ring in this way, including nitro, alkyl, acyl, and sulfonyl groups. Each proceeds via a similar mechanism (via a reactive electrophile that is generated in the reaction) either by using a catalyst or by using very reactive reagents.
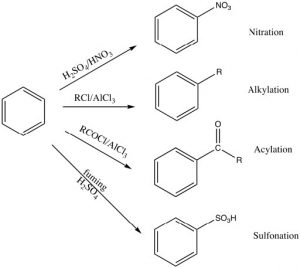
 Alkylation and acylation can be accomplished by treatment of an alkyl halide or acyl halide with a Lewis acid catalyst such as aluminum trichloride (AlCl3). In this case, the reactive electrophile is generated when the alkyl (or acyl) halide interacts with the catalyst to produce an intermediate that is carbocation-like. It is this very reactive species that reacts with the benzene ring to produce the substituted benzene. This reaction is called a Friedel-Crafts alkylation (or acylation). The acylation reaction is usually preferred because it is difficult to stop an alkylation reaction at just one substitution since the product is more reactive than the starting material (see below).
Alkylation and acylation can be accomplished by treatment of an alkyl halide or acyl halide with a Lewis acid catalyst such as aluminum trichloride (AlCl3). In this case, the reactive electrophile is generated when the alkyl (or acyl) halide interacts with the catalyst to produce an intermediate that is carbocation-like. It is this very reactive species that reacts with the benzene ring to produce the substituted benzene. This reaction is called a Friedel-Crafts alkylation (or acylation). The acylation reaction is usually preferred because it is difficult to stop an alkylation reaction at just one substitution since the product is more reactive than the starting material (see below).
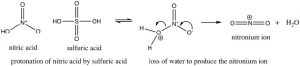
Nitration is accomplished by treating benzene with a mixture of concentrated nitric and sulfuric acids. This mixture generates a nitronium ion (NO2+), which is the reactive electrophile.
Sulfonation is accomplished by using fuming sulfuric acid, which actually contains sulfur trioxide (SO3) dissolved in the sulfuric acid. It is the actually the SO3 that is the electrophile in this case.
Some groups cannot be introduced directly onto the ring: for example, groups that we might normally consider as nucleophiles (such as NH2 or OH) have to be introduced indirectly. For example, aniline (aminobenzene) can be produced from nitrobenzene by reduction.
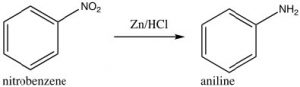
As we will see later, other nucleophilic groups have to be introduced by a different approach.
Multiple Substituents: Directing Effects.
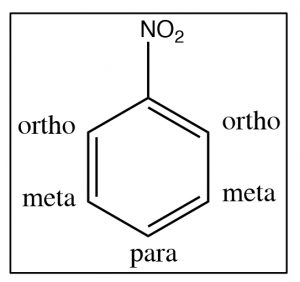 In this section, we will consider how substituents on the ring affect further reactions. It turns out that we can classify substituents into three groups. To understand and identify what these classifications are, let us take a look at some evidence beginning with nitrobenzene. There are two ways to identify positions on a substituted benzene ring: one way is simply to number the ring starting at the substituent; the other (which can only be used for disubstituted benzenes) is the ortho, meta, and para nomenclature as shown [latex]\rightarrow[/latex].
In this section, we will consider how substituents on the ring affect further reactions. It turns out that we can classify substituents into three groups. To understand and identify what these classifications are, let us take a look at some evidence beginning with nitrobenzene. There are two ways to identify positions on a substituted benzene ring: one way is simply to number the ring starting at the substituent; the other (which can only be used for disubstituted benzenes) is the ortho, meta, and para nomenclature as shown [latex]\rightarrow[/latex].
As we have seen, nitrobenzene can be produced by treating benzene with a mixture of nitric and sulfuric acids. If this reaction is carried out at room temperature, nitrobenzene is produced. If the reaction mixture is heated, eventually another product is produced. While there are three possible products (1,2- or 1,3- or 1,4-dinitrobenzene) only 1,3-dinitrobenzene (or meta-nitrobenzene) is formed. In addition, it is formed much more slowly and it is quite easy to isolate only the mono-nitro product.
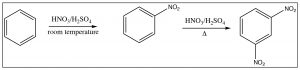
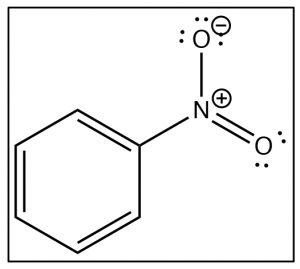 To understand why only one further product is formed and why the rate of the second substitution is slower (so that it requires more energy to surmount the activation energy barrier), let’s take a look at nitrobenzene itself. Notice that the positively charged nitrogen is adjacent to the ring. We might expect that this would remove some electron density from the ring and indeed it does.
To understand why only one further product is formed and why the rate of the second substitution is slower (so that it requires more energy to surmount the activation energy barrier), let’s take a look at nitrobenzene itself. Notice that the positively charged nitrogen is adjacent to the ring. We might expect that this would remove some electron density from the ring and indeed it does.
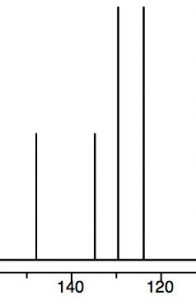 We can see this effect both in the C-13 and 1H NMR spectra, where the peaks are shifted to the lower field. Recall that benzene H signals all appear at 7.3 while in nitrobenzene we see that not only are there different signals, but that they are all at a lower field than benzene. There are chemically distinguishable hydrogens in nitrobenzene (2 ortho, 2 meta and 1 para).
We can see this effect both in the C-13 and 1H NMR spectra, where the peaks are shifted to the lower field. Recall that benzene H signals all appear at 7.3 while in nitrobenzene we see that not only are there different signals, but that they are all at a lower field than benzene. There are chemically distinguishable hydrogens in nitrobenzene (2 ortho, 2 meta and 1 para).
The lowest field 2H doublet at 8.2 ppm corresponds to the ortho Hs, the 2H signal at 7.6 ppm is the two meta Hs, and the signal at 7.5 is the para H. In the C-13 NMR spectrum, all the carbons in benzene appear at 128 ppm. In nitrobenzene, C-1, the carbon bonded to the nitro group is, as we might expect, shifted downfield to 148 ppm because of its proximity to the positively charged nitrogen.
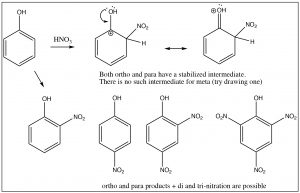
The evidence shows us that nitrobenzene is less electron-rich than benzene—in fact, it reacts with electrophiles 10,000 times more slowly than benzene—but why does it only produce one product on dinitration? The answer to this lies in the structure of the intermediate sigma complex. If you draw out the resonance forms for reaction at the ortho, meta, and para positions, what becomes clear is that the meta position is the only one that does not (cannot) place the negative charge on the original nitro-substituted carbon. Both ortho and para substitution are highly disfavored because these intermediates are of such high energy (because of the adjacent positive charges), and, therefore, only the meta product is formed.
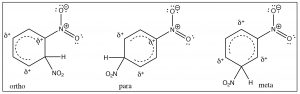
Nitro groups belong to the class of substituents that are deactivating meta directors. Other substitutents that belong in this group are sulfonic acids (SO3H) and any group with a carbonyl next to the ring (aldehydes, ketones, carboxylic acids, and derivatives). All of these groups are destabilized by having a positive charge on the adjacent carbon.
It makes sense then that there is another group of substituents that are activating ortho, para directors. For example, all alkyl groups fall into this classification because they are electron donating by induction. Toluene (methyl benzene) is the simplest example, and indeed when we nitrate toluene, we see a mixture of ortho and para products because now these are the intermediate sigma complexes that can be directly stabilized by induction from the methyl group.
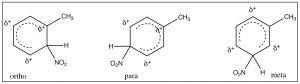
In this case, the ortho and para substitution products are stabilized and produced preferentially. The ratios of products are approximately 2:1 ortho to para (because there are two possible ortho positions). Indeed toluene can be nitrated further until all the ortho and para positions are substituted to give 2,4,6-trinitrotoluene, otherwise known as TNT.[8]
Another group of ortho, para directors are substituents that are attached to the ring by an O or N (that is, phenol) alkoxybenzenes or aniline and its derivatives. While it might seem, at first glance, that these electronegative atoms would be electron-withdrawing by induction, we know that they can also donate electrons through resonance with the ring. 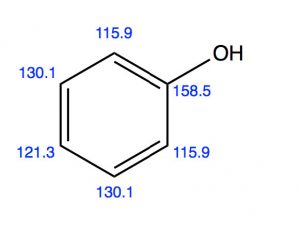 We can see both modes in operation if we look at the evidence from the C-13 NMR. C-1 appears at a much lower field (158 ppm) than benzene (128ppm), presumably because it is directly attached to the electron-withdrawing O. But the ortho and para carbons appear at a slightly higher field than benzene because these positions are enriched in electron density by resonance.
We can see both modes in operation if we look at the evidence from the C-13 NMR. C-1 appears at a much lower field (158 ppm) than benzene (128ppm), presumably because it is directly attached to the electron-withdrawing O. But the ortho and para carbons appear at a slightly higher field than benzene because these positions are enriched in electron density by resonance.
In fact, phenol is more reactive than benzene and it is difficult to stop the reaction at the mono-nitration step. Again, we observe a mixture of ortho and para products, because the transition state on the way to the intermediate sigma complex can be stabilized by resonance.
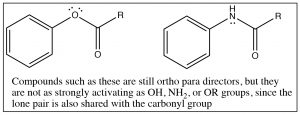
Generally, any benzene derivatives with a lone pair that is available for stabilizing the intermediate will be an ortho, para director including aniline (and any N-alkylated analogs) and anisole (methoxybenzene). This also includes compounds that have a lone pair that must be shared between the ring and a carbonyl group: for example, phenyl esters or amides.
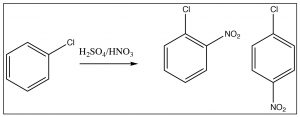
There is one more classification for directing groups: the halogens are anomalous in that they are deactivating ortho, para directors. Halogens are electronegative and withdraw electrons from the ring by induction, thus reducing the reactivity. However, halogens also have lone pairs that can be used to stabilize positive charge by resonance. The resonance effect is not as large as it is for N- or O-substituted rings, but it can still operate. Therefore halo-substituted benzene rings tend to react more slowly than benzene, but they do produce ortho and para products.
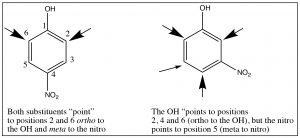
Multiple substituents: While it is relatively easy to predict the position of substitution on a monosubstituted ring (one you know how to do), predicting the outcome when there are two or more often becomes problematic. Sometimes, both substituents will “point” to the same positions. For example, in p-nitrophenol, both the OH and NO2 direct the substituent to the same position, but in m-nitrophenol, they direct to different positions as shown.
It is, therefore, sometimes possible to predict the position of the third substituent, but, often, it is not. Regardless of this problem, most aromatic substitution reactions have the potential for producing multiple products, either because both ortho and para products are formed or because the product may be more reactive than the reactant. For example, any electrophilic aromatic substitution that adds an activating group to the ring may be difficult to stop at one substitution. Friedel Crafts alkylation is difficult to stop at only one alkylation. In fact, rather than alkylate the ring, it is often preferable to acylate the ring, followed by a reduction of the acyl group. There are a number of reducing reagents that can take the aldehyde or ketone right down to an alkyl group: for example, as shown here, the Clemmensen reduction involves a zinc-mercury amalgam in HCl.
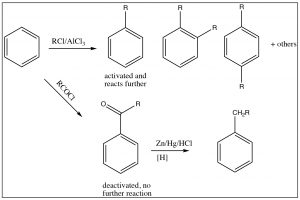
Nucleophilic Substitutions on Aromatic Systems: Expanding the range of potential substitution products.
All of the reactions on the aromatic ring so far have proceeded by one mechanism: electrophilic aromatic substitution. 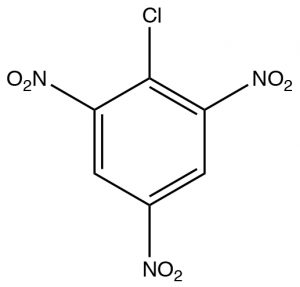 Up to now, we have not solved the problem of introducing substituents that usually react as nucleophiles onto an aromatic ring. There are a number of ways to accomplish this and we will consider three of these mechanisms.
Up to now, we have not solved the problem of introducing substituents that usually react as nucleophiles onto an aromatic ring. There are a number of ways to accomplish this and we will consider three of these mechanisms.
The first, Nucleophilic Aromatic Substitution (SNAr), is somewhat analogous to an SN2 reaction. It will occur if two conditions are met: the first is there must be a leaving group present on the ring, typically a halide; the second is that that the electron density of the ring must be reduced. This is typically accomplished by having electron-withdrawing groups such as nitro groups on the ring. We can observe the effect of adding electron-withdrawing groups by looking at the NMR spectra of 2-chloro-1,3,5-trinitrobenzene. The only peak in the H-NMR spectrum is a 2H singlet at 9.1 ppm. This is considerably downfield of the 7.3 ppm of benzene because the of the effect of the electron withdrawing nitro groups. This effect explains why the ring is susceptible to nucleophilic attack.
The reaction proceeds by an initial attack by a nucleophile such as OH, OR, followed by loss of the leaving group which takes the pair of electrons with it.
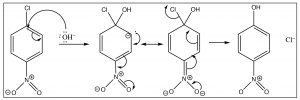
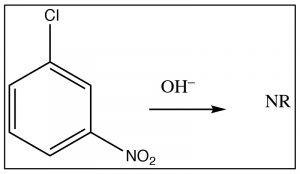 The intermediate anion can be directly stabilized by the nitro group, but only if that group is in the ortho or para position. If the nitro group is meta to the leaving group the reaction will not occur, because such stabilization is not possible (try to draw resonance forms for it).
The intermediate anion can be directly stabilized by the nitro group, but only if that group is in the ortho or para position. If the nitro group is meta to the leaving group the reaction will not occur, because such stabilization is not possible (try to draw resonance forms for it).
A second mechanism involves elimination of HX from the ring, followed by a rapid addition of HY; it can be considered analogous to elimination and addition reactions of alkenes and alkynes. The first step is the elimination of HX from benzene, which produces a highly reactive, strained species which is called a benzyne. This intermediate undergoes very rapid reaction to add H+ and Y– across the double bond.
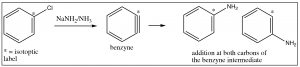
Evidence for this mechanism, rather than SNAr, comes from isotope labeling studies. If the original site is isotopically labeled (e.g. with C-13), the final product has only 50% of the nucleophile at the labeled site and 50% at the adjacent carbon. If this were a straight nucleophilic substitution, all of the substitution would take place at the labeled carbon.
Diazonium ions:
Another approach that allows access to multiple products involves the reaction of aniline (PhNH2) with the nitronium ion (produced in the reaction mixture) to what is known as a diazonium ion.
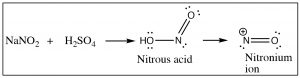
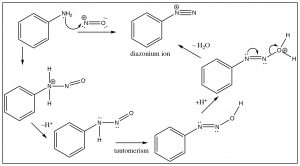
The diazonium ion is highly unstable: it must be prepared at temperatures around 0°C; if the solution is warmed above this temperature, it will decompose by losing a molecule of nitrogen (N2) to produce a carbocation. It is this decomposition that can be captured by a range of nucleophiles. 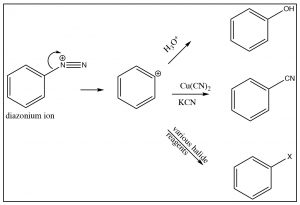 In some ways this reaction is akin to an SN1 reaction in which a carbocation is produced, which then undergoes rapid nucleophilic attack.
In some ways this reaction is akin to an SN1 reaction in which a carbocation is produced, which then undergoes rapid nucleophilic attack.
The intermediate carbocation can be captured in the presence of a nucleophile: for example water, alcohol, halide, or cyanide ions. Note that all these reactions typically require a raised temperature or the presence of a metal ion catalyst. This is to enhance the rate of decomposition of the diazonium ion, so that the resulting carbocation can be captured by the nucleophile. It is important to note that this carbocation is NOT resonance-stabilized (you cannot draw resonance forms to stabilize—try it).
Diazo coupling reactions: Besides being a way to introduce some nucleophiles into aromatic rings, diazonium salts also undergo what is known as a coupling reaction. The diazonium ion itself is susceptible to nucleophilic attack if it is not decomposed too rapidly—usually by another aromatic system. This reaction has been used to produce a wide range of dyes and indicators.
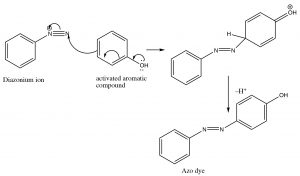
These azo compounds have a long-conjugated chromophore that typically results in absorption of visible light, making the compounds highly colored (as a result of the light that is reflected and not absorbed). Manipulation of groups on either benzene ring can extend the conjugation even further. About 50% of dyes belong to this family of compounds.
Reactions of Substituted Benzenes: Reaction at the Benzylic Position.
The position next to the benzene ring is special because reactive species such as carbanions, carbocations, or radicals at that site can be conjugated (and therefore stabilized) with the benzene pi system. For example, we have already seen that phenol (C6H5OH, pKa ~10) is much more acidic than a typical alcohol (pKa ~16) because the negative charge can be stabilized in the aromatic ring. Similarly, benzyl halides ( e.e. C6H5CH2Br) undergo nucleophilic substitution under SN1 conditions, even though these compounds appear to be primary halides, a carbocation next to the ring can be stabilized.
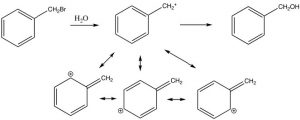
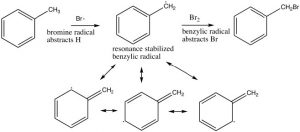
The position next to the ring is called the benzylic position, and is particularly reactive because of the ability to stabilize any intermediate in the aromatic ring. For example, it is possible to selectively introduce a bromine at the benzylic position via a reaction in which radicals are generated. We have spent little time on the reactions of organic compounds with radicals[9], because, generally, these reactions are very difficult to control. For example, an alkane will react with halogens in the presence of light or peroxides (which initiate the reaction by forming a radical), but the reaction is not synthetically useful and typically the halogen can end up in all the possible positions.
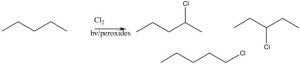
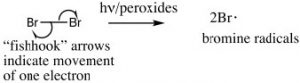 However, we can selectively introduce a bromine atom at a benzylic position, because the intermediate benzylic radical is most stable, and will have a lower activation energy to formation. The reaction begins by producing a bromine radical from Br2 by breaking the bond with light to give two Br radicals. The Br radical abstracts (removes) an H from the benzylic position to give the resonance-stabilized benzylic radical which then abstracts a Br from bromine (Br2).
However, we can selectively introduce a bromine atom at a benzylic position, because the intermediate benzylic radical is most stable, and will have a lower activation energy to formation. The reaction begins by producing a bromine radical from Br2 by breaking the bond with light to give two Br radicals. The Br radical abstracts (removes) an H from the benzylic position to give the resonance-stabilized benzylic radical which then abstracts a Br from bromine (Br2).
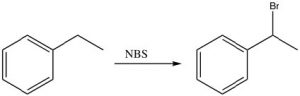
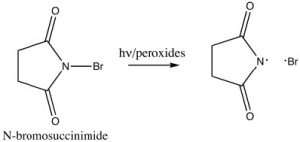 In practice, we use a source of bromine radicals that is easier to handle than elemental bromine, N-bromosuccinimide (NBS), which has a weak N-Br bond that will break homolytically (i.e. to give two radicals) in the presence of light or peroxides. NBS will react with alkyl benzenes to introduce a bromine specifically at the benzylic position.
In practice, we use a source of bromine radicals that is easier to handle than elemental bromine, N-bromosuccinimide (NBS), which has a weak N-Br bond that will break homolytically (i.e. to give two radicals) in the presence of light or peroxides. NBS will react with alkyl benzenes to introduce a bromine specifically at the benzylic position.
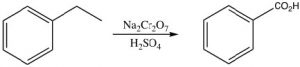
Another unique reaction of benzylic positions is that they can be oxidized by reagents such as KMnO4 to give the corresponding carboxylic acid; any other carbons in the side chain are removed in the process.
Pericyclic reactions:
 Up to this point, we have focused on two types of reactions that can be broadly classified as Lewis acid-base (electrophile-nucleophile) reactions and, to a lesser extent, on oxidation-reduction reactions. Both of these reaction types are typically initiated by differences in electron density—that is, interactions between molecules or parts of molecules that are electrostatically interacting with each other.
Up to this point, we have focused on two types of reactions that can be broadly classified as Lewis acid-base (electrophile-nucleophile) reactions and, to a lesser extent, on oxidation-reduction reactions. Both of these reaction types are typically initiated by differences in electron density—that is, interactions between molecules or parts of molecules that are electrostatically interacting with each other. 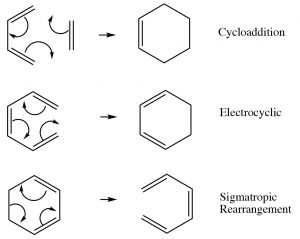 They may occur via ionic or radical intermediates. However, there is a different type of reaction that is not initiated in this way; instead the reaction occurs via a cyclic movement of electrons, breaking and forming bonds in a concerted fashion (at the same time). These reactions are called pericyclic reactions and can occur with a variety of substrates. The three major types of these reactions are shown[latex]\rightarrow[/latex]. What is interesting about them is that they involve the cyclic rearrangement of six electrons.
They may occur via ionic or radical intermediates. However, there is a different type of reaction that is not initiated in this way; instead the reaction occurs via a cyclic movement of electrons, breaking and forming bonds in a concerted fashion (at the same time). These reactions are called pericyclic reactions and can occur with a variety of substrates. The three major types of these reactions are shown[latex]\rightarrow[/latex]. What is interesting about them is that they involve the cyclic rearrangement of six electrons.
Diels-Alder Reactions:
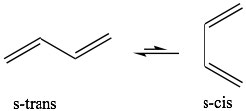
The best-known of these reactions is a cycloaddition reaction known as the Diels-Alder reaction. 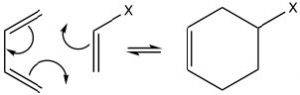 It typically occurs between a conjugated diene and another alkene known as the dienophile. This reaction is very useful in synthesis because it forms two new C-C bonds at the same time. It is also stereospecific and regiospecific. The simplest diene that can participate in a Diels-Alder is butadiene. Typically, butadiene tends to exist in the more stable s-trans[10] conformation—however, there is an equilibrium concentration of the s-cis conformation through which the reaction can occur. In fact, cyclopentadiene, in which the diene is permanently fused in the s-trans conformation, reacts rapidly with itself (it can be both diene and dienophile) in a reversible reaction.
It typically occurs between a conjugated diene and another alkene known as the dienophile. This reaction is very useful in synthesis because it forms two new C-C bonds at the same time. It is also stereospecific and regiospecific. The simplest diene that can participate in a Diels-Alder is butadiene. Typically, butadiene tends to exist in the more stable s-trans[10] conformation—however, there is an equilibrium concentration of the s-cis conformation through which the reaction can occur. In fact, cyclopentadiene, in which the diene is permanently fused in the s-trans conformation, reacts rapidly with itself (it can be both diene and dienophile) in a reversible reaction.
The “dienophile” can be a simple alkene, but the presence of an electron-withdrawing group (such as a carbonyl group) on the double bond improves yields.  We can envision the reaction as taking place in a concerted fashion. Again, the reaction is reversible, and the reverse reaction is known as a retro Diels-Alder. We can use our knowledge of thermodynamics to predict the most appropriate conditions for the reaction. Recall that the extent of any reaction can be predicted from the Gibbs free-energy change ΔG = ΔH –TΔS. Since the reaction produces two new C-C single bonds and one new C-C pi bond while breaking two C-C pi bonds, we can assume that the enthalpy change for this reaction is negative (bond formation releases energy and bond-breaking uses energy). Therefore, the ΔH term is always favorable. We can also predict the sign of the entropy change for the system; since we are producing one molecule from two, we would expect ΔS to be negative also. The entropy term is unfavorable. From this analysis, we can see that the temperature at which the reaction is carried out is crucial. High temperatures would favor the reverse reaction. Therefore Diels-Alder reactions are typically run at fairly moderate temperatures that are between room temperature and 150°C).
We can envision the reaction as taking place in a concerted fashion. Again, the reaction is reversible, and the reverse reaction is known as a retro Diels-Alder. We can use our knowledge of thermodynamics to predict the most appropriate conditions for the reaction. Recall that the extent of any reaction can be predicted from the Gibbs free-energy change ΔG = ΔH –TΔS. Since the reaction produces two new C-C single bonds and one new C-C pi bond while breaking two C-C pi bonds, we can assume that the enthalpy change for this reaction is negative (bond formation releases energy and bond-breaking uses energy). Therefore, the ΔH term is always favorable. We can also predict the sign of the entropy change for the system; since we are producing one molecule from two, we would expect ΔS to be negative also. The entropy term is unfavorable. From this analysis, we can see that the temperature at which the reaction is carried out is crucial. High temperatures would favor the reverse reaction. Therefore Diels-Alder reactions are typically run at fairly moderate temperatures that are between room temperature and 150°C).
The possibilities for Diels-Alder reactions are quite extensive and since this is a concerted reaction, 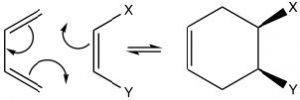 any stereochemistry in the starting materials is conserved in the products. For example, if the dienophile has cis or trans stereochemistry, this is conserved in the product. The cis dienophile gives the cis product (and vice versa for the trans).
any stereochemistry in the starting materials is conserved in the products. For example, if the dienophile has cis or trans stereochemistry, this is conserved in the product. The cis dienophile gives the cis product (and vice versa for the trans).
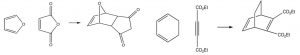
Some examples of Diels-Alder reactions are given here.
While we can draw mechanistic arrows for pericyclic reactions such as Diels-Alder, they are best understood by using molecular orbital theory. In this treatment, we can consider the reaction as taking place between the highest occupied molecular orbital (HOMO) of the diene and the lowest unoccupied molecular orbital (LUMO) of the dienophile.
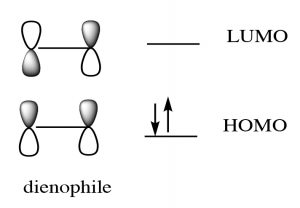
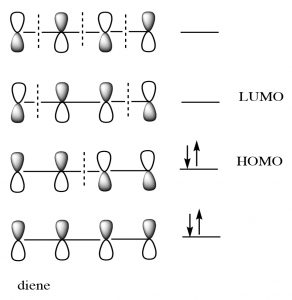
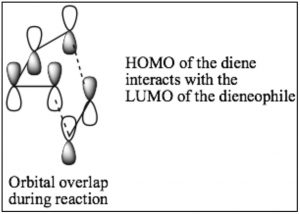 For our purposes, this treatment is too complex, but if you go on to further studies of pericyclic reactions, MO theory will be the approach that will allow you to predict the outcome of many different reactions.
For our purposes, this treatment is too complex, but if you go on to further studies of pericyclic reactions, MO theory will be the approach that will allow you to predict the outcome of many different reactions.
If a fused ring is formed during the reaction, there are two possibilities for the orientation of the substituents on the diene as shown below: exo and endo.
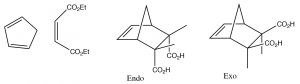
Because of the possibility of a stabilizing interaction in the endo position, (the pi system of the carboxylic acid can interact with the pi system of the diene), the endo product is usually produced.
As noted earlier, there are other types of pericyclic reactions. All of the common reactions that occur simply by heating up the starting materials involve the cyclic movement of six electrons. The transition states for these reactions all involve molecular orbitals that extend throughout the system; these orbitals have considerably lower energy than one might expect. In some ways, this is analogous to the 6 pi electrons of aromatic systems, which are also stabilized in lower energy molecular orbitals.
Interestingly, these reactions do not occur in the same way if they are initiated by electromagnetic radiation (that is, if we shine light on them rather than heat them up). In this case the electrons that participate in the reaction are actually in higher energy orbitals (the electron absorbs a photon and is promoted to a higher energy level). Absorbing light leads to a completely different set of reactions and outcomes, something that is explored in subsequent organic chemistry courses. There is, however, one particularly interesting (and biologically relevant) photochemically induced reaction, namely the reaction of adjacent thymine bases within a DNA molecule. Upon the absorption of a UV photon, such adjacent thymines can undergo an electrocyclic reaction that results in dimer formation.
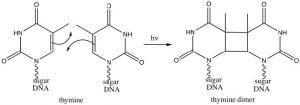
The presence of a thymine dimer results in conformational changes in the DNA that, unrepaired, can lead to mutations. Thymine dimers are recognized and repaired via two distinct cellular-repair mechanisms. Unrepaired damage can lead to skin cancer (a range of carcinomas and melanomas).
This is one reason to limit skin exposure to the sun.
- Recall that this is how relative stabilities of alkenes were determined (Chapter 4). ↵
- A longer version of this brief overview is found in Chapter 2. ↵
- http://phototroph.blogspot.com/2006/11/pigments-and-absorption-spectra.html ↵
- Recall we saw this same tautomerism when water adds across a triple bond. ↵
- https://en.wikipedia.org/wiki/Gilman_reagent ↵
- The explanation for this phenomenon goes beyond the scope of this course and is best explained using the theory of hard and soft acids and bases. For more information see https://en.wikipedia.org/wiki/HSAB_theory ↵
- This is in contrast to simple hydrocarbons which do not smell. In fact, methane thiol (CH3SH) must be added to methane and propane which are used for heating so that they can be detected in the event of a gas leak. ↵
- TNT is an explosive compound, as are many nitrated organic compounds (for example nitroglycerin). The nitro group is relatively unstable (NO bonds are weak) and these compounds can decompose explosively to produce more stable nitrogen oxides and CO2, releasing a great amount of energy at the same time. https://en.wikipedia.org/wiki/Trinitrotoluene ↵
- Except for the radical-induced addition of HBr in an anti-Markovinkov manner across a double bond. ↵
- Recall that there is some double bond character between carbons 2 and 3 and, therefore, rotation is somewhat restricted around this bond unlike a normal C-C single bond. ↵
